Nucleophilic describes the affinity of a nucleophile to bond with positively charged atomic nuclei. Nucleophilicity, sometimes referred to as nucleophile strength, refers to a substance's nucleophilic character and is often used to compare the affinity of atoms. Neutral nucleophilic reactions with solvents such as alcohols and water are named solvolysis. Nucleophiles may take part in nucleophilic substitution, whereby a nucleophile becomes attracted to a full or partial positive charge, and nucleophilic addition. Nucleophilicity is closely related to basicity. The difference between the two is, that basicity is a thermodynamic property (i.e. relates to an equilibrium state), but nucleophilicity is a kinetic property, which relates to rates of certain chemical reactions.[1]
History and etymology
The terms nucleophile and electrophile were introduced by Christopher Kelk Ingold in 1933,[2] replacing the terms anionoid and cationoid proposed earlier by A. J. Lapworth in 1925.[3] The word nucleophile is derived from nucleus and the Greek word φιλος, philos, meaning friend.[4][5]
Properties
In general, in a group across the periodic table, the more basic the ion (the higher the pKa of the conjugate acid) the more reactive it is as a nucleophile. Within a series of nucleophiles with the same attacking element (e.g. oxygen), the order of nucleophilicity will follow basicity. Sulfur is in general a better nucleophile than oxygen.[citation needed]
Nucleophilicity
Many schemes attempting to quantify relative nucleophilic strength have been devised. The following empirical data have been obtained by measuring reaction rates for many reactions involving many nucleophiles and electrophiles. Nucleophiles displaying the so-called alpha effect are usually omitted in this type of treatment.[citation needed]
Swain–Scott equation
The first such attempt is found in the Swain–Scott equation[6][7] derived in 1953:
This free-energy relationship relates the pseudo first orderreaction rate constant (in water at 25°C), k, of a reaction, normalized to the reaction rate, k0, of a standard reaction with water as the nucleophile, to a nucleophilic constant n for a given nucleophile and a substrate constant s that depends on the sensitivity of a substrate to nucleophilic attack (defined as 1 for methyl bromide).
The Ritchie equation, derived in 1972, is another free-energy relationship:[8][9][10]
where N+ is the nucleophile dependent parameter and k0 the reaction rate constant for water. In this equation, a substrate-dependent parameter like s in the Swain–Scott equation is absent. The equation states that two nucleophiles react with the same relative reactivity regardless of the nature of the electrophile, which is in violation of the reactivity–selectivity principle. For this reason, this equation is also called the constant selectivity relationship.
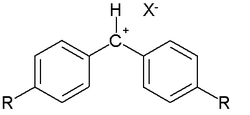
In the original publication the data were obtained by reactions of selected nucleophiles with selected electrophilic carbocations such as tropylium or diazonium cations:
or (not displayed) ions based on malachite green. Many other reaction types have since been described.
The second orderreaction rate constantk at 20°C for a reaction is related to a nucleophilicity parameter N, an electrophilicity parameter E, and a nucleophile-dependent slope parameter s. The constant s is defined as 1 with 2-methyl-1-pentene as the nucleophile.
Typical N values with s in parentheses are −4.47 (1.32) for electrophilic aromatic substitution to toluene (1), −0.41 (1.12) for electrophilic addition to 1-phenyl-2-propene (2), and 0.96 (1) for addition to 2-methyl-1-pentene (3), −0.13 (1.21) for reaction with triphenylallylsilane (4), 3.61 (1.11) for reaction with 2-methylfuran (5), +7.48 (0.89) for reaction with isobutenyltributylstannane (6) and +13.36 (0.81) for reaction with the enamine 7.[13]
With E = −9.15 for the S-methyldibenzothiophenium ion, typical nucleophile values N (s) are 15.63 (0.64) for piperidine, 10.49 (0.68) for methoxide, and 5.20 (0.89) for water. In short, nucleophilicities towards sp2 or sp3 centers follow the same pattern.
Unified equation
In an effort to unify the above described equations the Mayr equation is rewritten as:[14]
with sE the electrophile-dependent slope parameter and sN the nucleophile-dependent slope parameter. This equation can be rewritten in several ways:
with sE = 1 for carbocations this equation is equal to the original Mayr–Patz equation of 1994,
with sN = 0.6 for most n nucleophiles the equation becomes
or the original Scott–Swain equation written as:
with sE = 1 for carbocations and sN = 0.6 the equation becomes:
or the original Ritchie equation written as:
Types
Examples of nucleophiles are anions such as Cl−, or a compound with a lone pair of electrons such as NH3 (ammonia) and PR3.[citation needed]
In the example below, the oxygen of the hydroxide ion donates an electron pair to form a new chemical bond with the carbon at the end of the bromopropane molecule. The bond between the carbon and the bromine then undergoes heterolytic fission, with the bromine atom taking the donated electron and becoming the bromide ion (Br−), because a SN2 reaction occurs by backside attack. This means that the hydroxide ion attacks the carbon atom from the other side, exactly opposite the bromine ion. Because of this backside attack, SN2 reactions result in an inversion of the configuration of the electrophile. If the electrophile is chiral, it typically maintains its chirality, though the SN2 product's absolute configuration is flipped as compared to that of the original electrophile.[citation needed]
Ambident nucleophile
An ambident nucleophile is one that can attack from two or more places, resulting in two or more products. For example, the thiocyanate ion (SCN−) may attack from either the sulfur or the nitrogen. For this reason, the SN2 reaction of an alkyl halide with SCN− often leads to a mixture of an alkyl thiocyanate (R-SCN) and an alkyl isothiocyanate (R-NCS). Similar considerations apply in the Kolbe nitrile synthesis.[citation needed]
Halogens
While the halogens are not nucleophilic in their diatomic form (e.g. I2 is not a nucleophile), their anions are good nucleophiles. In polar, protic solvents, F− is the weakest nucleophile, and I− the strongest; this order is reversed in polar, aprotic solvents.[15]
Of sulfur nucleophiles, hydrogen sulfide and its salts, thiols (RSH), thiolate anions (RS−), anions of thiolcarboxylic acids (RC(O)-S−), and anions of dithiocarbonates (RO-C(S)-S−) and dithiocarbamates (R2N-C(S)-S−) are used most often.
In general, sulfur is very nucleophilic because of its large size, which makes it readily polarizable, and its lone pairs of electrons are readily accessible.
Although metal centers (e.g., Li+, Zn2+, Sc3+, etc.) are most commonly cationic and electrophilic (Lewis acidic) in nature, certain metal centers (particularly ones in a low oxidation state and/or carrying a negative charge) are among the strongest recorded nucleophiles and are sometimes referred to as "supernucleophiles." For instance, using methyl iodide as the reference electrophile, Ph3Sn– is about 10000 times more nucleophilic than I–, while the Co(I) form of vitamin B12 (vitamin B12s) is about 107 times more nucleophilic.[16] Other supernucleophilic metal centers include low oxidation state carbonyl metalate anions (e.g., CpFe(CO)2–).[17]
Examples
The following table shows the nucleophilicity of some molecules with methanol as the solvent:[18]
Relative nucleophilicity
Molecules
Very Good
I⁻, HS⁻, RS⁻
Good
Br⁻, OH⁻, RO⁻, CN⁻, N3⁻
Fair
NH3, Cl⁻, F⁻, RCO2⁻
Weak
H2O, ROH
Very Weak
RCO2H
See also
Electrophile– Chemical species that accepts an electron pair from a nucleophile
↑Ingold, C. K. (1933). "266. Significance of tautomerism and of the reactions of aromatic compounds in the electronic theory of organic reactions". Journal of the Chemical Society (Resumed): 1120. doi:10.1039/jr9330001120.
↑Lapworth, A. (1925). "Replaceability of Halogen Atoms by Hydrogen Atoms". Nature. 115: 625.
↑Mayr, Herbert; Patz, Matthias (1994). "Scales of Nucleophilicity and Electrophilicity: A System for Ordering Polar Organic and Organometallic Reactions". Angewandte Chemie International Edition in English. 33 (9): 938. doi:10.1002/anie.199409381.
↑Mayr, Herbert; Bug, Thorsten; Gotta, Matthias F; Hering, Nicole; Irrgang, Bernhard; Janker, Brigitte; Kempf, Bernhard; Loos, Robert; Ofial, Armin R; Remennikov, Grigoriy; Schimmel, Holger (2001). "Reference Scales for the Characterization of Cationic Electrophiles and Neutral Nucleophiles". Journal of the American Chemical Society. 123 (39): 9500–12. Bibcode:2001JAChS.123.9500M. doi:10.1021/ja010890y. PMID11572670. S2CID8392147.
↑Dessy, Raymond E.; Pohl, Rudolph L.; King, R. Bruce (November 1966). "Organometallic Electrochemistry. VII. 1 The Nucleophilicities of Metallic and Metalloidal Anions Derived from Metals of Groups IV, V, VI, VII, and VIII". Journal of the American Chemical Society. 88 (22): 5121–5124. Bibcode:1966JAChS..88.5121D. doi:10.1021/ja00974a015. ISSN0002-7863.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.