Related Research Articles
Computational chemistry is a branch of chemistry that uses computer simulation to assist in solving chemical problems. It uses methods of theoretical chemistry, incorporated into computer programs, to calculate the structures and properties of molecules, groups of molecules, and solids. It is essential because, apart from relatively recent results concerning the hydrogen molecular ion, the quantum many-body problem cannot be solved analytically, much less in closed form. While computational results normally complement the information obtained by chemical experiments, it can in some cases predict hitherto unobserved chemical phenomena. It is widely used in the design of new drugs and materials.

Molecular mechanics uses classical mechanics to model molecular systems. The Born–Oppenheimer approximation is assumed valid and the potential energy of all systems is calculated as a function of the nuclear coordinates using force fields. Molecular mechanics can be used to study molecule systems ranging in size and complexity from small to large biological systems or material assemblies with many thousands to millions of atoms.
MOLPRO is a software package used for accurate ab initio quantum chemistry calculations. It is developed by Peter Knowles at Cardiff University and Hans-Joachim Werner at Universität Stuttgart in collaboration with other authors.
Gaussian is a general purpose computational chemistry software package initially released in 1970 by John Pople and his research group at Carnegie Mellon University as Gaussian 70. It has been continuously updated since then. The name originates from Pople's use of Gaussian orbitals to speed up molecular electronic structure calculations as opposed to using Slater-type orbitals, a choice made to improve performance on the limited computing capacities of then-current computer hardware for Hartree–Fock calculations. The current version of the program is Gaussian 16. Originally available through the Quantum Chemistry Program Exchange, it was later licensed out of Carnegie Mellon University, and since 1987 has been developed and licensed by Gaussian, Inc.
Q-Chem is a general-purpose electronic structure package featuring a variety of established and new methods implemented using innovative algorithms that enable fast calculations of large systems on various computer architectures, from laptops and regular lab workstations to midsize clusters and HPCC, using density functional and wave-function based approaches. It offers an integrated graphical interface and input generator; a large selection of functionals and correlation methods, including methods for electronically excited states and open-shell systems; solvation models; and wave-function analysis tools. In addition to serving the computational chemistry community, Q-Chem also provides a versatile code development platform.
In chemistry, a hypervalent molecule is a molecule that contains one or more main group elements apparently bearing more than eight electrons in their valence shells. Phosphorus pentachloride, sulfur hexafluoride, chlorine trifluoride, the chlorite ion, and the triiodide ion are examples of hypervalent molecules.
In the context of chemistry and molecular modelling, a force field is a computational method that is used to estimate the forces between atoms within molecules and also between molecules. More precisely, the force field refers to the functional form and parameter sets used to calculate the potential energy of a system of atoms or coarse-grained particles in molecular mechanics, molecular dynamics, or Monte Carlo simulations. The parameters for a chosen energy function may be derived from experiments in physics and chemistry, calculations in quantum mechanics, or both. Force fields are interatomic potentials and utilize the same concept as force fields in classical physics, with the difference that the force field parameters in chemistry describe the energy landscape, from which the acting forces on every particle are derived as a gradient of the potential energy with respect to the particle coordinates.

PQS is a general purpose quantum chemistry program. Its roots go back to the first ab initio gradient program developed in Professor Peter Pulay's group but now it is developed and distributed commercially by Parallel Quantum Solutions. There is a reduction in cost for academic users and a site license. Its strong points are geometry optimization, NMR chemical shift calculations, and large MP2 calculations, and high parallel efficiency on computing clusters. It includes many other capabilities including Density functional theory, the semiempirical methods, MINDO/3, MNDO, AM1 and PM3, Molecular mechanics using the SYBYL 5.0 Force Field, the quantum mechanics/molecular mechanics mixed method using the ONIOM method, natural bond orbital (NBO) analysis and COSMO solvation models. Recently, a highly efficient parallel CCSD(T) code for closed shell systems has been developed. This code includes many other post Hartree–Fock methods: MP2, MP3, MP4, CISD, CEPA, QCISD and so on.

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
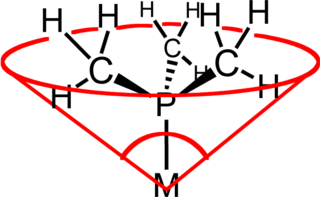
In coordination chemistry, the ligand cone angle is a measure of the steric bulk of a ligand in a transition metal coordination complex. It is defined as the solid angle formed with the metal at the vertex and the outermost edge of the van der Waals spheres of the ligand atoms at the perimeter of the cone. Tertiary phosphine ligands are commonly classified using this parameter, but the method can be applied to any ligand. The term cone angle was first introduced by Chadwick A. Tolman, a research chemist at DuPont. Tolman originally developed the method for phosphine ligands in nickel complexes, determining them from measurements of accurate physical models.
Free energy perturbation (FEP) is a method based on statistical mechanics that is used in computational chemistry for computing free energy differences from molecular dynamics or Metropolis Monte Carlo simulations.
Physical organic chemistry, a term coined by Louis Hammett in 1940, refers to a discipline of organic chemistry that focuses on the relationship between chemical structures and reactivity, in particular, applying experimental tools of physical chemistry to the study of organic molecules. Specific focal points of study include the rates of organic reactions, the relative chemical stabilities of the starting materials, reactive intermediates, transition states, and products of chemical reactions, and non-covalent aspects of solvation and molecular interactions that influence chemical reactivity. Such studies provide theoretical and practical frameworks to understand how changes in structure in solution or solid-state contexts impact reaction mechanism and rate for each organic reaction of interest.
The hybrid QM/MM approach is a molecular simulation method that combines the strengths of ab initio QM calculations (accuracy) and MM (speed) approaches, thus allowing for the study of chemical processes in solution and in proteins. The QM/MM approach was introduced in the 1976 paper of Warshel and Levitt. They, along with Martin Karplus, won the 2013 Nobel Prize in Chemistry for "the development of multiscale models for complex chemical systems".
Quantum chemistry composite methods are computational chemistry methods that aim for high accuracy by combining the results of several calculations. They combine methods with a high level of theory and a small basis set with methods that employ lower levels of theory with larger basis sets. They are commonly used to calculate thermodynamic quantities such as enthalpies of formation, atomization energies, ionization energies and electron affinities. They aim for chemical accuracy which is usually defined as within 1 kcal/mol of the experimental value. The first systematic model chemistry of this type with broad applicability was called Gaussian-1 (G1) introduced by John Pople. This was quickly replaced by the Gaussian-2 (G2) which has been used extensively. The Gaussian-3 (G3) was introduced later.
An electronic effect influences the structure, reactivity, or properties of molecule but is neither a traditional bond nor a steric effect. In organic chemistry, the term stereoelectronic effect is also used to emphasize the relation between the electronic structure and the geometry (stereochemistry) of a molecule.
CP2K is a freely available (GPL) quantum chemistry and solid state physics program package, written in Fortran 2008, to perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems. It provides a general framework for different methods: density functional theory (DFT) using a mixed Gaussian and plane waves approach (GPW) via LDA, GGA, MP2, or RPA levels of theory, classical pair and many-body potentials, semi-empirical and tight-binding Hamiltonians, as well as Quantum Mechanics/Molecular Mechanics (QM/MM) hybrid schemes relying on the Gaussian Expansion of the Electrostatic Potential (GEEP). The Gaussian and Augmented Plane Waves method (GAPW) as an extension of the GPW method allows for all-electron calculations. CP2K can do simulations of molecular dynamics, metadynamics, Monte Carlo, Ehrenfest dynamics, vibrational analysis, core level spectroscopy, energy minimization, and transition state optimization using NEB or dimer method.

Newton-X is a general program for molecular dynamics simulations beyond the Born-Oppenheimer approximation. It has been primarily used for simulations of ultrafast processes in photoexcited molecules. It has also been used for simulation of band envelops of absorption and emission spectra.

Marcin Maciej Hoffmann is a Polish scientist and entrepreneur. He is a professor of chemistry at the Faculty of Chemistry of Adam Mickiewicz University in Poznań.
Dispersion stabilized molecules are molecules where the London dispersion force (LDF), a non-covalent attractive force between atoms and molecules, plays a significant role in promoting the molecule's stability. Distinct from steric hindrance, dispersion stabilization has only recently been considered in depth by organic and inorganic chemists after earlier gaining prominence in protein science and supramolecular chemistry. Although usually weaker than covalent bonding and other forms of non-covalent interactions like hydrogen bonding, dispersion forces are known to be a significant if not dominating stabilizing force in certain organic, inorganic, and main group molecules, stabilizing otherwise reactive moieties and exotic bonding.
References
- ↑ "Investigating the Reactivity and Spectra of Large Molecules with ONIOM | Gaussian.com". gaussian.com. Retrieved 2023-04-13.
- ↑ S. Dapprich; I. Komaromi; K.S. Byun; K. Morokuma & M.J. Frisch (1999). "A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives". Journal of Molecular Structure: THEOCHEM. 461–462: 1–21. doi:10.1016/S0166-1280(98)00475-8.
- ↑ Vreven, T; Morokuma, K (2006). "Chapter 3 Hybrid Methods: ONIOM(QM:MM) and QM/MM". Annual Reports in Computational Chemistry. 2: 35–51. doi:10.1016/S1574-1400(06)02003-2. ISBN 9780444528223.
- ↑ Svensson, Mats; Humbel, StéPhane; Froese, Robert D. J.; Matsubara, Toshiaki; Sieber, Stefan; Morokuma, Keiji (1996). "ONIOM: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels−Alder Reactions and Pt(P(t-Bu)3)2+ H2Oxidative Addition". The Journal of Physical Chemistry. 100 (50): 19357. doi:10.1021/jp962071j.
- ↑ Senn, H; Thiel, W (2007). "QM/MM studies of enzymes". Current Opinion in Chemical Biology. 11 (2): 182–7. doi:10.1016/j.cbpa.2007.01.684. PMID 17307018.
- ↑ Ananikov, Valentine P.; Musaev, Djamaladdin G.; Morokuma, Keiji (2010). "Real size of ligands, reactants and catalysts: Studies of structure, reactivity and selectivity by ONIOM and other hybrid computational approaches☆". Journal of Molecular Catalysis A: Chemical. 324 (1–2): 104–119. doi:10.1016/j.molcata.2010.03.015.
| | This quantum chemistry-related article is a stub. You can help Wikipedia by expanding it. |