Ceric ammonium nitrate (CAN) is the inorganic compound with the formula (NH4)2Ce(NO3)6. This orange-red, water-soluble cerium salt is a specialised oxidizing agent in organic synthesis and a standard oxidant in quantitative analysis.

Pyridinium chlorochromate (PCC) is a yellow-orange salt with the formula [C5H5NH]+[CrO3Cl]−. It is a reagent in organic synthesis used primarily for oxidation of alcohols to form carbonyls. A variety of related compounds are known with similar reactivity. PCC offers the advantage of the selective oxidation of alcohols to aldehydes or ketones, whereas many other reagents are less selective.
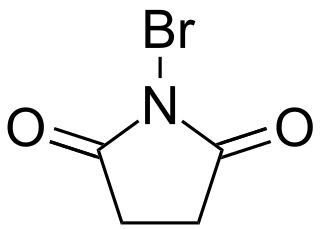
N-Bromosuccinimide or NBS is a chemical reagent used in radical substitution, electrophilic addition, and electrophilic substitution reactions in organic chemistry. NBS can be a convenient source of Br•, the bromine radical.

Dess–Martin periodinane (DMP) is a chemical reagent used in the Dess–Martin oxidation, oxidizing primary alcohols to aldehydes and secondary alcohols to ketones. This periodinane has several advantages over chromium- and DMSO-based oxidants that include milder conditions, shorter reaction times, higher yields, simplified workups, high chemoselectivity, tolerance of sensitive functional groups, and a long shelf life. However, use on an industrial scale is made difficult by its cost and its potentially explosive nature. It is named after the American chemists Daniel Benjamin Dess and James Cullen Martin who developed the reagent in 1983. It is based on IBX, but due to the acetate groups attached to the central iodine atom, DMP is much more reactive than IBX and is much more soluble in organic solvents.

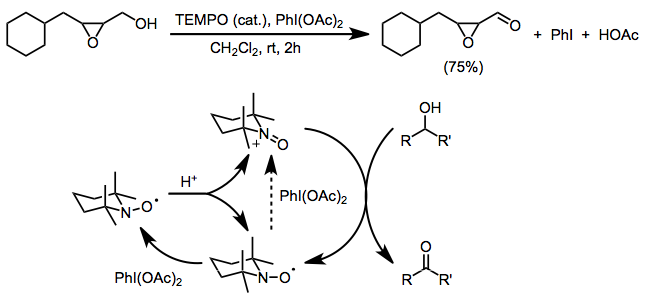
N-Oxoammonium salts are a class of organic compounds with the formula [R1R2+N=O]X−. The cation [R1R2+N=O] is of interest for the dehydrogenation of alcohols. Oxoammonium salts are diamagnetic, whereas the nitroxide has a doublet ground state. A prominent nitroxide is prepared by oxidation of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl, commonly referred to as [TEMPO]+. A less expensive analogue is Bobbitt's salt.
Dihydroxylation is the process by which an alkene is converted into a vicinal diol. Although there are many routes to accomplish this oxidation, the most common and direct processes use a high-oxidation-state transition metal. The metal is often used as a catalyst, with some other stoichiometric oxidant present. In addition, other transition metals and non-transition metal methods have been developed and used to catalyze the reaction.
Oppenauer oxidation, named after Rupert Viktor Oppenauer, is a gentle method for selectively oxidizing secondary alcohols to ketones.

The Cornforth reagent or pyridinium dichromate(PDC) is a pyridinium salt of dichromate with the chemical formula [C5H5NH]2[Cr2O7]. This compound is named after the Australian-British chemist Sir John Warcup Cornforth (b. 1917) who introduced it in 1962. The Cornforth reagent is a strong oxidizing agent which can convert primary and secondary alcohols to aldehydes and ketones respectively. In its chemical structure and functions it is closely related to other compounds made from hexavalent chromium oxide, such as pyridinium chlorochromate and Collins reagent. Because of their toxicity, these reagents are rarely used nowadays.
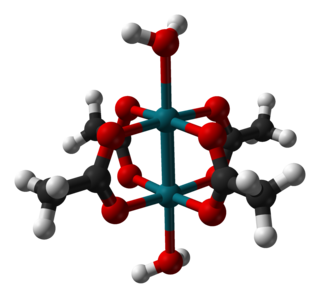
Rhodium(II) acetate is the coordination compound with the formula Rh2(AcO)4, where AcO− is the acetate ion (CH
3CO−
2). This dark green powder is slightly soluble in polar solvents, including water. It is used as a catalyst for cyclopropanation of alkenes. It is a widely studied example of a transition metal carboxylate complex.

The oxidation of primary alcohols to carboxylic acids is an important oxidation reaction in organic chemistry.
Alcohol oxidation is a class of organic reactions in which the alcohol functional group is converted into another functional group in which carbon carries a higher oxidation state.
Oxidation with chromium(VI) complexes involves the conversion of alcohols to carbonyl compounds or more highly oxidized products through the action of molecular chromium(VI) oxides and salts. The principal reagents are Collins reagent, PDC, and PCC. These reagents represent improvements over inorganic chromium(VI) reagents such as Jones reagent.
Oxidation with dioxiranes refers to the introduction of oxygen into organic molecules through the action of a dioxirane. Dioxiranes are well known for their oxidation of alkenes to epoxides; however, they are also able to oxidize other unsaturated functionality, heteroatoms, and alkane C-H bonds.
Nucleophilic epoxidation is the formation of epoxides from electron-deficient double bonds through the action of nucleophilic oxidants. Nucleophilic epoxidation methods represent a viable alternative to electrophilic methods, many of which do not epoxidize electron-poor double bonds efficiently.
Manganese-mediated coupling reactions are radical coupling reactions between enolizable carbonyl compounds and unsaturated compounds initiated by a manganese(III) salt, typically manganese(III) acetate. Copper(II) acetate is sometimes used as a co-oxidant to assist in the oxidation of intermediate radicals to carbocations.

(2,2,6,6-Tetramethylpiperidin-1-yl)oxyl or (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl, commonly known as TEMPO, is a chemical compound with the formula (CH2)3(CMe2)2NO. This heterocyclic compound is a red-orange, sublimable solid. As a stable aminoxyl radical, it has applications in chemistry and biochemistry. TEMPO is used as a radical marker, as a structural probe for biological systems in conjunction with electron spin resonance spectroscopy, as a reagent in organic synthesis, and as a mediator in controlled radical polymerization.

James McCue Bobbitt was an American chemist and academic who taught chemistry at the University of Connecticut from 1956 to 1991 and developed the Bobbitt reaction.
Sulfonium-based oxidations of alcohols to aldehydes summarizes a group of organic reactions that transform a primary alcohol to the corresponding aldehyde (and a secondary alcohol to the corresponding ketone). Selective oxidation of alcohols to aldehydes requires circumventing over-oxidation to the carboxylic acid. One popular approach are methods that proceed through intermediate alkoxysulfonium species (RO−SMe+
2X-, e.g. compound 6) as detailed here. Since most of these methods employ dimethylsulfoxide (DMSO) as oxidant and generate dimethylsulfide, these are often colloquially summarized as DMSO-oxidations. Conceptually, generating an aldehyde and dimethylsulfide from an alcohol and DMSO requires a dehydrating agent for removal of H2O, ideally an electrophile simultaneously activating DMSO. In contrast, methods generating the sulfonium intermediate from dimethylsulfide do not require a dehydrating agent. Closely related are oxidations mediated by dimethyl selenoxide and by dimethyl selenide.
The Stahl oxidation is a copper-catalyzed aerobic oxidation of primary and secondary alcohols to aldehydes and ketones. Known for its high selectivity and mild reaction conditions, the Stahl oxidation offers several advantages over classical alcohol oxidations.

The Babler oxidation, also known as the Babler-Dauben oxidation, is an organic reaction for the oxidative transposition of tertiary allylic alcohols to enones using pyridinium chlorochromate (PCC):