
Epidermolytic ichthyosis (EI), is a rare and severe form of ichthyosis that affects around 1 in 300,000 people. It is caused by a genetic mutation, and thus cannot be completely cured without some form of gene therapy.
Type II keratins constitutes the Type II intermediate filaments (IFs) of the intracytoplasmatic cytoskeleton, which is present in all mammalian epithelial cells. The type 2 cytokeratins consist of basic or neutral, high molecular weight proteins which in vivo are arranged in pairs of heterotypic Type I and Type II keratin chains, coexpressed during differentiation of simple and stratified epithelial tissues. It has been seen that Type II Keratins are developed before Type 1 keratins during human embryonic development.

Keratin 16 is a protein that in humans is encoded by the KRT16 gene.

Palmoplantar keratodermas are a heterogeneous group of disorders characterized by abnormal thickening of the stratum corneum of the palms and soles.

Ectodermal dysplasia (ED) is a group of genetic syndromes all deriving from abnormalities of the ectodermal structures. More than 150 different syndromes have been identified.

Hyperkeratosis is thickening of the stratum corneum, often associated with the presence of an abnormal quantity of keratin, and also usually accompanied by an increase in the granular layer. As the corneum layer normally varies greatly in thickness in different sites, some experience is needed to assess minor degrees of hyperkeratosis.

Meleda disease (MDM) or "mal de Meleda", also called Mljet disease, keratosis palmoplantaris and transgradiens of Siemens, is an extremely rare autosomal recessive congenital skin disorder in which dry, thick patches of skin develop on the soles of the hands and feet, a condition known as palmoplantar hyperkeratosis. Meleda Disease is a skin condition which usually can be identified not long after birth. This is a genetic condition but it is very rare. The hands and feet usually are the first to show signs of the disease but the disease can advance to other parts of the body. Signs of the disease include thickening of the skin, on hands and soles of feet, which can turn red in color. There currently is no cure and treatment is limited, but Acitretin can be used in severe cases.

Papillon–Lefèvre syndrome (PLS), also known as palmoplantar keratoderma with periodontitis, is an autosomal recessive genetic disorder caused by a deficiency in cathepsin C.

Dermatopathia pigmentosa reticularis(DPR) is a rare, autosomal dominant congenital disorder that is a form of ectodermal dysplasia. Dermatopathia pigmentosa reticularis is composed of the triad of generalized reticulate hyperpigmentation, noncicatricial alopecia, and onychodystrophy. DPR is a non life-threatening disease that largely affects the skin, hair, and nails. It has also been identified as a keratin disorder. Historically, as of 1992, only 10 cases had been described in world literature; however, due to recent advances in genetic analysis, five additional families studied in 2006 have been added to the short list of confirmed cases.
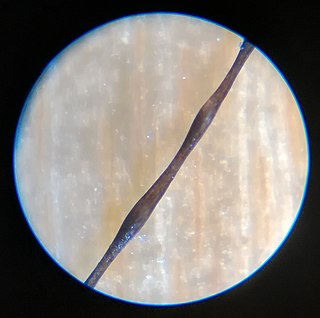
Monilethrix is a rare autosomal dominant hair disease that results in short, fragile, broken hair that appears beaded. It comes from the Latin word for necklace (monile) and the Greek word for hair (thrix). Hair becomes brittle, and breaks off at the thinner parts between the beads. It appears as a thinning or baldness of hair and was first described in 1897 by Walter Smith

Keratin 6C, is a type II cytokeratin, one of a number of isoforms of keratin 6 encoded by separate genes located within the type II keratin gene cluster on human chromosome 12q. This gene was uncovered recently by the Human Genome Project and its expression patterns in humans remains unknown.

Genodermatosis is a hereditary skin disease with three inherited modes including single gene inheritance, multiple gene inheritance and chromosome inheritance. There are many different types of genodermatosis, the prevalence of genodermatosis ranges from 1 per 6000 people to 1 per 500,000 people. Genodermatosis has influence on the texture, color and structure of skin cuticle and connective tissue, specific lesion site and clinical manifestations on the body vary depending on the type. In the spite of the variety and complexity of genodermatosis, there are still some common methods that can help people diagnose. After diagnosis, different types of genodermatosis require different levels of therapy including interventions, nursing interventions and treatments. Among that, research of therapy for some new, complex and rare types are still in the developing stage. The impact of genodermatosis not only can be seen in body but also can be seen in all aspects of patients' life, including but not limited to psychological, family life, economic conditions and social activities. Accordingly, the patients need treatment, support and help in these areas.

Steatocystoma multiplex, is a benign, autosomal dominant congenital condition resulting in multiple cysts on a person's body. Steatocystoma simplex is the solitary counterpart to steatocystoma multiplex.

Ichthyosis bullosa of Siemens is a type of familial, autosomal dominant ichthyosis, a rare skin disorder. It is also known as bullous congenital ichthyosiform erythroderma of Siemens or ichthyosis exfoliativa. It is a genetic disorder with no known cure which is estimated to affect about 1 in 500,000 people.
Peeling skin syndrome is an autosomal recessive disorder characterized by lifelong peeling of the stratum corneum, and may be associated with pruritus, short stature, and easily removed anagen hair.
Schöpf–Schulz–Passarge syndrome is an autosomal recessive condition with punctate symmetric palmoplantar keratoderma, with the keratoderma and fragility of the nails beginning around age 12. In addition to palmoplantar keratoderma, other symptoms include hypodontia, hypotrichosis, nail dystrophies, and eyelid cysts. Patients may also develop syringofibroadenoma and squamous cell carcinomas.
Howel–Evans syndrome is an extremely rare condition involving thickening of the skin in the palms of the hands and the soles of the feet (hyperkeratosis). This familial disease is associated with a high lifetime risk of esophageal cancer. For this reason, it is sometimes known as tylosis with oesophageal cancer (TOC).
Haim–Munk syndrome is a skin disease caused, like Papillon–Lefèvre syndrome, by a mutation in the cathepsin C gene. One of its features is thick curved finger and toenails.
Palmoplantar keratoderma with deafness, also known as Palmoplantar keratoderma-deafness syndrome is a rare genetic disorder which is characterized by either focal or diffuse early-onset palmoplantar keratoderma and sensorineural deafness. Transmission is autosomal dominant with incomplete penetrance.
