
Huntington's disease (HD), also known as Huntington's chorea, is an incurable neurodegenerative disease that is mostly inherited. The earliest symptoms are often subtle problems with mood or mental/psychiatric abilities. A general lack of coordination and an unsteady gait often follow. It is also a basal ganglia disease causing a hyperkinetic movement disorder known as chorea. As the disease advances, uncoordinated, involuntary body movements of chorea become more apparent. Physical abilities gradually worsen until coordinated movement becomes difficult and the person is unable to talk. Mental abilities generally decline into dementia, depression, apathy, and impulsivity at times. The specific symptoms vary somewhat between people. Symptoms usually begin between 30 and 50 years of age, and can start at any age but are usually seen around the age of 40. The disease may develop earlier in each successive generation. About eight percent of cases start before the age of 20 years, and are known as juvenile HD, which typically present with the slow movement symptoms of Parkinson's disease rather than those of chorea.

A frameshift mutation is a genetic mutation caused by indels of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame, resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein. A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.

Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a neurological condition in its own right. An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. SCA is hereditary, progressive, degenerative, and often fatal. There is no known effective treatment or cure. SCA can affect anyone of any age. The disease is caused by either a recessive or dominant gene. In many cases people are not aware that they carry a relevant gene until they have children who begin to show signs of having the disorder.
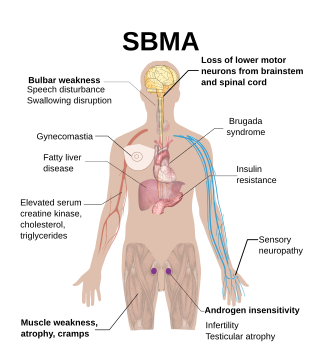
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.
In genetics, trinucleotide repeat disorders, a subset of microsatellite expansion diseases, are a set of over 30 genetic disorders caused by trinucleotide repeat expansion, a kind of mutation in which repeats of three nucleotides increase in copy numbers until they cross a threshold above which they cause developmental, neurological or neuromuscular disorders. In addition to the expansions of these trinucleotide repeats, expansions of one tetranucleotide (CCTG), five pentanucleotide, three hexanucleotide, and one dodecanucleotide (CCCCGCCCCGCG) repeat cause 13 other diseases. Depending on its location, the unstable trinucleotide repeat may cause defects in a protein encoded by a gene; change the regulation of gene expression; produce a toxic RNA, or lead to production of a toxic protein. In general, the larger the expansion the faster the onset of disease, and the more severe the disease becomes.

Huntingtin(Htt) is the protein coded for in humans by the HTT gene, also known as the IT15 ("interesting transcript 15") gene. Mutated HTT is the cause of Huntington's disease (HD), and has been investigated for this role and also for its involvement in long-term memory storage.
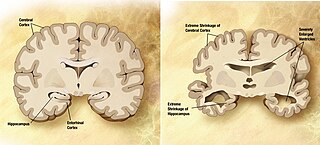
A neurodegenerative disease is caused by the progressive loss of neurons, in the process known as neurodegeneration. Neuronal damage may also ultimately result in their death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

Ataxin-1 is a DNA-binding protein which in humans is encoded by the ATXN1 gene.
Ataxin 7 (ATXN7) is a protein of the SCA7 gene, which contains 892 amino acids with an expandable poly(Q) region close to the N-terminus. The expandable poly(Q) motif region in the protein contributes crucially to spinocerebellar ataxia (SCA) pathogenesis by the induction of intranuclear inclusion bodies. ATXN7 is associated with both olivopontocerebellar atrophy type 3 (OPCA3) and spinocerebellar ataxia type 7 (SCA7).
Ataxin is a type of nuclear protein. The class is called ataxin because mutated forms of these proteins and their corresponding genes were found to cause progressive ataxia.

Spinocerebellar ataxia type 6 (SCA6) is a rare, late-onset, autosomal dominant disorder, which, like other types of SCA, is characterized by dysarthria, oculomotor disorders, peripheral neuropathy, and ataxia of the gait, stance, and limbs due to cerebellar dysfunction. Unlike other types, SCA 6 is not fatal. This cerebellar function is permanent and progressive, differentiating it from episodic ataxia type 2 (EA2) where said dysfunction is episodic. In some SCA6 families, some members show these classic signs of SCA6 while others show signs more similar to EA2, suggesting that there is some phenotypic overlap between the two disorders. SCA6 is caused by mutations in CACNA1A, a gene encoding a calcium channel α subunit. These mutations tend to be trinucleotide repeats of CAG, leading to the production of mutant proteins containing stretches of 20 or more consecutive glutamine residues; these proteins have an increased tendency to form intracellular agglomerations. Unlike many other polyglutamine expansion disorders expansion length is not a determining factor for the age that symptoms present.

Ataxin-2 is a protein that in humans is encoded by the ATXN2 gene. Mutations in ATXN2 cause spinocerebellar ataxia type 2 (SCA2).

Ataxin-3 is a protein that in humans is encoded by the ATXN3 gene.

Junctophilin-3 (JPH3) is a protein residing in humans that is encoded by the JPH3 gene. The gene is approximately 97 kilobases long and is located at chromosomal position 16q24.2. Junctophilin proteins are associated with the formation of junctional membrane complexes, which link the plasma membrane with the endoplasmic reticulum in excitable cells. JPH3 is localized to the brain and is associated with motor coordination and memory neurons.
Dentatorubral–pallidoluysian atrophy (DRPLA) is an autosomal dominant spinocerebellar degeneration caused by an expansion of a CAG repeat encoding a polyglutamine tract in the atrophin-1 protein. It is also known as Haw River Syndrome and Naito–Oyanagi disease. Although this condition was perhaps first described by Smith et al. in 1958, and several sporadic cases have been reported from Western countries, this disorder seems to be very rare except in Japan.
A polyglutamine tract or polyQ tract is a portion of a protein consisting of a sequence of several glutamine units. A tract typically consists of about 10 to a few hundred such units.
Autosomal dominant cerebellar ataxia (ADCA) is a form of spinocerebellar ataxia inherited in an autosomal dominant manner. ADCA is a genetically inherited condition that causes deterioration of the nervous system leading to disorder and a decrease or loss of function to regions of the body.
Spinocerebellar ataxia type 1 (SCA1) is a rare autosomal dominant disorder, which, like other spinocerebellar ataxias, is characterized by neurological symptoms including dysarthria, hypermetric saccades, and ataxia of gait and stance. This cerebellar dysfunction is progressive and permanent. First onset of symptoms is normally between 30 and 40 years of age, though juvenile onset can occur. Death typically occurs within 10 to 30 years from onset.

Chromosome 9 open reading frame 43 is a protein that in humans is encoded by the C9orf43 gene. The gene is also known as MGC17358 and LOC257169. C9orf43 contains DUF 4647 and a polyglutamine repeat region although protein function is not well understood.

Nancy M. Bonini is an American neuroscientist and geneticist, best known for pioneering the use of Drosophila as a model organism to study neurodegeneration of the human brain. Using the Drosophila model approach, Bonini's laboratory has identified genes and pathways that are important in the development and progression of neurodegenerative diseases such as Amyotrophic lateral sclerosis, Alzheimer's disease, and Parkinson's disease, as well as aging, neural injury and regeneration, and response to environmental toxins.