
Polymerase chain reaction (PCR) is a method widely used to rapidly make millions to billions of copies of a specific DNA sample, allowing scientists to take a very small sample of DNA and amplify it to a large enough amount to study in detail. PCR was invented in 1983 by the American biochemist Kary Mullis at Cetus Corporation; Mullis and biochemist Michael Smith, who had developed other essential ways of manipulating DNA, were jointly awarded the Nobel Prize in Chemistry in 1993.

A primer is a short single-stranded nucleic acid used by all living organisms in the initiation of DNA synthesis. DNA polymerase enzymes are only capable of adding nucleotides to the 3’-end of an existing nucleic acid, requiring a primer be bound to the template before DNA polymerase can begin a complementary strand. DNA polymerase adds nucleotides after binding to the RNA primer and synthesis the whole strand. Later, the RNA strands must be removed accurately and replace them with DNA nucleotides forming a gap region known as a nick that is filled in using an enzyme called ligase. The removal process of the RNA primer requires several enzymes, such as Fen1, Lig1, and others that work in coordination with DNA polymerase, to ensure the removal of the RNA nucleotides and the addition of DNA nucleotides. Living organisms use solely RNA primers, while laboratory techniques in biochemistry and molecular biology that require in vitro DNA synthesis usually use DNA primers, since they are more temperature stable. Primers can be designed in laboratory for specific reactions such as polymerase chain reaction (PCR). When designing PCR primers, there are specific measures that must be taken into consideration, like the melting temperature of the primers and the annealing temperature of the reaction itself. Moreover, the DNA binding sequence of the primer in vitro has to be specifically chosen, which is done using a method called basic local alignment search tool (BLAST) that scans the DNA and finds specific and unique regions for the primer to bind.
Protein engineering is the process of developing useful or valuable proteins. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It is also a product and services market, with an estimated value of $168 billion by 2017.
Site-directed mutagenesis is a molecular biology method that is used to make specific and intentional mutating changes to the DNA sequence of a gene and any gene products. Also called site-specific mutagenesis or oligonucleotide-directed mutagenesis, it is used for investigating the structure and biological activity of DNA, RNA, and protein molecules, and for protein engineering.
A DNA construct is an artificially-designed segment of DNA borne on a vector that can be used to incorporate genetic material into a target tissue or cell. A DNA construct contains a DNA insert, called a transgene, delivered via a transformation vector which allows the insert sequence to be replicated and/or expressed in the target cell. This gene can be cloned from a naturally occurring gene, or synthetically constructed. The vector can be delivered using physical, chemical or viral methods. Typically, the vectors used in DNA constructs contain an origin of replication, a multiple cloning site, and a selectable marker. Certain vectors can carry additional regulatory elements based on the expression system involved.
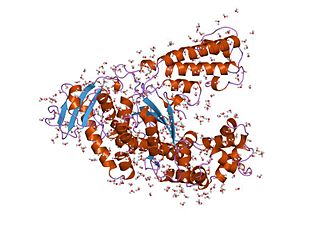
Taq polymerase is a thermostable DNA polymerase I named after the thermophilic eubacterial microorganism Thermus aquaticus, from which it was originally isolated by Chien et al. in 1976. Its name is often abbreviated to Taq or Taq pol. It is frequently used in the polymerase chain reaction (PCR), a method for greatly amplifying the quantity of short segments of DNA.
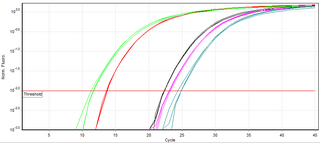
A real-time polymerase chain reaction is a laboratory technique of molecular biology based on the polymerase chain reaction (PCR). It monitors the amplification of a targeted DNA molecule during the PCR, not at its end, as in conventional PCR. Real-time PCR can be used quantitatively and semi-quantitatively.
The overlap extension polymerase chain reaction is a variant of PCR. It is also referred to as Splicing by overlap extension / Splicing by overhang extension (SOE) PCR. It is used to insert specific mutations at specific points in a sequence or to splice smaller DNA fragments into a larger polynucleotide.
TaqMan probes are hydrolysis probes that are designed to increase the specificity of quantitative PCR. The method was first reported in 1991 by researcher Kary Mullis at Cetus Corporation, and the technology was subsequently developed by Hoffmann-La Roche for diagnostic assays and by Applied Biosystems for research applications.
The polymerase chain reaction (PCR) is a commonly used molecular biology tool for amplifying DNA, and various techniques for PCR optimization which have been developed by molecular biologists to improve PCR performance and minimize failure.
Synthetic genomics is a nascent field of synthetic biology that uses aspects of genetic modification on pre-existing life forms, or artificial gene synthesis to create new DNA or entire lifeforms.

Artificial gene synthesis, or simply gene synthesis, refers to a group of methods that are used in synthetic biology to construct and assemble genes from nucleotides de novo. Unlike DNA synthesis in living cells, artificial gene synthesis does not require template DNA, allowing virtually any DNA sequence to be synthesized in the laboratory. It comprises two main steps, the first of which is solid-phase DNA synthesis, sometimes known as DNA printing. This produces oligonucleotide fragments that are generally under 200 base pairs. The second step then involves connecting these oligonucleotide fragments using various DNA assembly methods. Because artificial gene synthesis does not require template DNA, it is theoretically possible to make a completely synthetic DNA molecule with no limits on the nucleotide sequence or size.

Functional cloning is a molecular cloning technique that relies on prior knowledge of the encoded protein’s sequence or function for gene identification. In this assay, a genomic or cDNA library is screened to identify the genetic sequence of a protein of interest. Expression cDNA libraries may be screened with antibodies specific for the protein of interest or may rely on selection via the protein function. Historically, the amino acid sequence of a protein was used to prepare degenerate oligonucleotides which were then probed against the library to identify the gene encoding the protein of interest. Once candidate clones carrying the gene of interest are identified, they are sequenced and their identity is confirmed. This method of cloning allows researchers to screen entire genomes without prior knowledge of the location of the gene or the genetic sequence.

The history of the polymerase chain reaction (PCR) has variously been described as a classic "Eureka!" moment, or as an example of cooperative teamwork between disparate researchers. Following is a list of events before, during, and after its development:
The versatility of polymerase chain reaction (PCR) has led to a large number of variants of PCR.
Gibson assembly is a molecular cloning method which allows for the joining of multiple DNA fragments in a single, isothermal reaction. It is named after its creator, Daniel G. Gibson, who is the chief technology officer and co-founder of the synthetic biology company Codex DNA.

In silico PCR refers to computational tools used to calculate theoretical polymerase chain reaction (PCR) results using a given set of primers (probes) to amplify DNA sequences from a sequenced genome or transcriptome.
Clyde A. Hutchison III is an American biochemist and microbiologist notable for his research on site-directed mutagenesis and synthetic biology. He is Professor Emeritus of Microbiology and Immunology at the University of North Carolina at Chapel Hill, Distinguished Professor at the J Craig Venter Institute, a member of the National Academy of Sciences, and a fellow of the American Academy of Arts and Sciences.
Synthetic genome is a synthetically-built genome whose formation involves either genetic modification on pre-existing life forms or artificial gene synthesis to create new DNA or entire lifeforms. The field that studies synthetic genomes is called synthetic genomics.
SCAR-less genome editing Scarless Cas9 Assisted Recombineering (no-SCAR) is an editing method that is able to manipulate the Escherichia coli genome. The system relies on recombineering whereby DNA sequences are combined and manipulated through homologous recombination. No-SCAR is able to manipulate the E. coli genome without the use of the chromosomal markers detailed in previous recombineering methods. Instead, in this method, the λ-Red recombination system facilitates donor DNA integration while Cas9 cleaves double-stranded DNA to counter-select against wild-type cells. Although λ-Red and Cas9 genome editing are widely used technologies, the no-SCAR method is novel in combining the two functions; this technique is able to establish point mutations, gene deletions, and short sequence insertions in several genomic loci with increased efficiency and time sensitivity.
