Gene knockouts are a widely used genetic engineering technique that involves the targeted removal or inactivation of a specific gene within an organism's genome. This can be done through a variety of methods, including homologous recombination, CRISPR-Cas9, and TALENs.

Mosaicism or genetic mosaicism is a condition in which a multicellular organism possesses more than one genetic line as the result of genetic mutation. This means that various genetic lines resulted from a single fertilized egg. Mosaicism is one of several possible causes of chimerism, wherein a single organism is composed of cells with more than one distinct genotype.
Cre-Lox recombination is a site-specific recombinase technology, used to carry out deletions, insertions, translocations and inversions at specific sites in the DNA of cells. It allows the DNA modification to be targeted to a specific cell type or be triggered by a specific external stimulus. It is implemented both in eukaryotic and prokaryotic systems. The Cre-lox recombination system has been particularly useful to help neuroscientists to study the brain in which complex cell types and neural circuits come together to generate cognition and behaviors. NIH Blueprint for Neuroscience Research has created several hundreds of Cre driver mouse lines which are currently used by the worldwide neuroscience community.
Site-specific recombinase technologies are genome engineering tools that depend on recombinase enzymes to replace targeted sections of DNA.
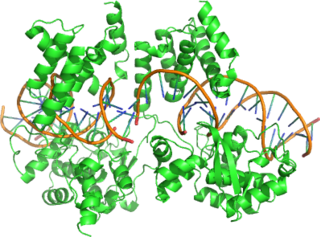
Cre recombinase is a tyrosine recombinase enzyme derived from the P1 bacteriophage. The enzyme uses a topoisomerase I-like mechanism to carry out site specific recombination events. The enzyme is a member of the integrase family of site specific recombinase and it is known to catalyse the site specific recombination event between two DNA recognition sites. This 34 base pair (bp) loxP recognition site consists of two 13 bp palindromic sequences which flank an 8bp spacer region. The products of Cre-mediated recombination at loxP sites are dependent upon the location and relative orientation of the loxP sites. Two separate DNA species both containing loxP sites can undergo fusion as the result of Cre mediated recombination. DNA sequences found between two loxP sites are said to be "floxed". In this case the products of Cre mediated recombination depends upon the orientation of the loxP sites. DNA found between two loxP sites oriented in the same direction will be excised as a circular loop of DNA whilst intervening DNA between two loxP sites that are opposingly orientated will be inverted. The enzyme requires no additional cofactors or accessory proteins for its function.
Recombineering is a genetic and molecular biology technique based on homologous recombination systems, as opposed to the older/more common method of using restriction enzymes and ligases to combine DNA sequences in a specified order. Recombineering is widely used for bacterial genetics, in the generation of target vectors for making a conditional mouse knockout, and for modifying DNA of any source often contained on a bacterial artificial chromosome (BAC), among other applications.

In genetics, Flp-FRT recombination is a site-directed recombination technology, increasingly used to manipulate an organism's DNA under controlled conditions in vivo. It is analogous to Cre-lox recombination but involves the recombination of sequences between short flippase recognition target (FRT) sites by the recombinase flippase (Flp) derived from the 2 µ plasmid of baker's yeast Saccharomyces cerevisiae.
P1 is a temperate bacteriophage that infects Escherichia coli and some other bacteria. When undergoing a lysogenic cycle the phage genome exists as a plasmid in the bacterium unlike other phages that integrate into the host DNA. P1 has an icosahedral head containing the DNA attached to a contractile tail with six tail fibers. The P1 phage has gained research interest because it can be used to transfer DNA from one bacterial cell to another in a process known as transduction. As it replicates during its lytic cycle it captures fragments of the host chromosome. If the resulting viral particles are used to infect a different host the captured DNA fragments can be integrated into the new host's genome. This method of in vivo genetic engineering was widely used for many years and is still used today, though to a lesser extent. P1 can also be used to create the P1-derived artificial chromosome cloning vector which can carry relatively large fragments of DNA. P1 encodes a site-specific recombinase, Cre, that is widely used to carry out cell-specific or time-specific DNA recombination by flanking the target DNA with loxP sites.
Site-specific recombination, also known as conservative site-specific recombination, is a type of genetic recombination in which DNA strand exchange takes place between segments possessing at least a certain degree of sequence homology. Enzymes known as site-specific recombinases (SSRs) perform rearrangements of DNA segments by recognizing and binding to short, specific DNA sequences (sites), at which they cleave the DNA backbone, exchange the two DNA helices involved, and rejoin the DNA strands. In some cases the presence of a recombinase enzyme and the recombination sites is sufficient for the reaction to proceed; in other systems a number of accessory proteins and/or accessory sites are required. Many different genome modification strategies, among these recombinase-mediated cassette exchange (RMCE), an advanced approach for the targeted introduction of transcription units into predetermined genomic loci, rely on SSRs.
The Tn3 transposon is a 4957 base pair mobile genetic element, found in prokaryotes. It encodes three proteins:

Gene targeting is a biotechnological tool used to change the DNA sequence of an organism. It is based on the natural DNA-repair mechanism of Homology Directed Repair (HDR), including Homologous Recombination. Gene targeting can be used to make a range of sizes of DNA edits, from larger DNA edits such as inserting entire new genes into an organism, through to much smaller changes to the existing DNA such as a single base-pair change. Gene targeting relies on the presence of a repair template to introduce the user-defined edits to the DNA. The user will design the repair template to contain the desired edit, flanked by DNA sequence corresponding (homologous) to the region of DNA that the user wants to edit; hence the edit is targeted to a particular genomic region. In this way Gene Targeting is distinct from natural homology-directed repair, during which the ‘natural’ DNA repair template of the sister chromatid is used to repair broken DNA. The alteration of DNA sequence in an organism can be useful in both a research context – for example to understand the biological role of a gene – and in biotechnology, for example to alter the traits of an organism.
Conditional gene knockout is a technique used to eliminate a specific gene in a certain tissue, such as the liver. This technique is useful to study the role of individual genes in living organisms. It differs from traditional gene knockout because it targets specific genes at specific times rather than being deleted from beginning of life. Using the conditional gene knockout technique eliminates many of the side effects from traditional gene knockout. In traditional gene knockout, embryonic death from a gene mutation can occur, and this prevents scientists from studying the gene in adults. Some tissues cannot be studied properly in isolation, so the gene must be inactive in a certain tissue while remaining active in others. With this technology, scientists are able to knockout genes at a specific stage in development and study how the knockout of a gene in one tissue affects the same gene in other tissues.
In molecular cloning and biology, a gene knock-in refers to a genetic engineering method that involves the one-for-one substitution of DNA sequence information in a genetic locus or the insertion of sequence information not found within the locus. Typically, this is done in mice since the technology for this process is more refined and there is a high degree of shared sequence complexity between mice and humans. The difference between knock-in technology and traditional transgenic techniques is that a knock-in involves a gene inserted into a specific locus, and is thus a "targeted" insertion. It is the opposite of gene knockout.
DNA adenine methyltransferase identification, often abbreviated DamID, is a molecular biology protocol used to map the binding sites of DNA- and chromatin-binding proteins in eukaryotes. DamID identifies binding sites by expressing the proposed DNA-binding protein as a fusion protein with DNA methyltransferase. Binding of the protein of interest to DNA localizes the methyltransferase in the region of the binding site. Adenine methylation does not occur naturally in eukaryotes and therefore adenine methylation in any region can be concluded to have been caused by the fusion protein, implying the region is located near a binding site. DamID is an alternate method to ChIP-on-chip or ChIP-seq.

In genetics, floxing refers to the sandwiching of a DNA sequence between two lox P sites. The terms are constructed upon the phrase "flanking/flanked by LoxP". Recombination between LoxP sites is catalysed by Cre recombinase. Floxing a gene allows it to be deleted, translocated or inverted in a process called Cre-Lox recombination. The floxing of genes is essential in the development of scientific model systems as it allows researchers to have spatial and temporal alteration of gene expression. Moreover, animals such as mice can be used as models to study human disease. Therefore, Cre-lox system can be used in mice to manipulate gene expression in order to study human diseases and drug development. For example, using the Cre-lox system, researchers can study oncogenes and tumor suppressor genes and their role in development and progression of cancer in mice models.

Genetic engineering techniques allow the modification of animal and plant genomes. Techniques have been devised to insert, delete, and modify DNA at multiple levels, ranging from a specific base pair in a specific gene to entire genes. There are a number of steps that are followed before a genetically modified organism (GMO) is created. Genetic engineers must first choose what gene they wish to insert, modify, or delete. The gene must then be isolated and incorporated, along with other genetic elements, into a suitable vector. This vector is then used to insert the gene into the host genome, creating a transgenic or edited organism.

Reverse genetics is a method in molecular genetics that is used to help understand the function(s) of a gene by analysing the phenotypic effects caused by genetically engineering specific nucleic acid sequences within the gene. The process proceeds in the opposite direction to forward genetic screens of classical genetics. While forward genetics seeks to find the genetic basis of a phenotype or trait, reverse genetics seeks to find what phenotypes are controlled by particular genetic sequences.
No-SCAR genome editing is an editing method that is able to manipulate the Escherichia coli genome. The system relies on recombineering whereby DNA sequences are combined and manipulated through homologous recombination. No-SCAR is able to manipulate the E. coli genome without the use of the chromosomal markers detailed in previous recombineering methods. Instead, the λ-Red recombination system facilitates donor DNA integration while Cas9 cleaves double-stranded DNA to counter-select against wild-type cells. Although λ-Red and Cas9 genome editing are widely used technologies, the no-SCAR method is novel in combining the two functions; this technique is able to establish point mutations, gene deletions, and short sequence insertions in several genomic loci with increased efficiency and time sensitivity.
Susan M. Dymecki is an American geneticist and neuroscientist and director of the Biological and Biomedical Sciences PhD Program at Harvard University. Dymecki is also a professor in the Department of Genetics and the principal investigator of the Dymecki Lab at Harvard. Her lab characterizes the development and function of unique populations of serotonergic neurons in the mouse brain. To enable this functional dissection, Dymecki has pioneered several transgenic tools for probing neural circuit development and function. Dymecki also competed internationally as an ice dancer, placing 7th in the 1980 U.S. Figure Skating Championships.

Genome editing of synthetic target arrays for lineage tracing (GESTALT) is a method used to determine the developmental lineages of cells in multicellular systems. GESTALT involves introducing a small DNA barcode that contains regularly spaced CRISPR/Cas9 target sites into the genomes of progenitor cells. Alongside the barcode, Cas9 and sgRNA are introduced into the cells. Mutations in the barcode accumulate during the course of cell divisions and the unique combination of mutations in a cell's barcode can be determined by DNA or RNA sequencing to link it to a developmental lineage.

