In organic chemistry, an alkane, or paraffin, is an acyclic saturated hydrocarbon. In other words, an alkane consists of hydrogen and carbon atoms arranged in a tree structure in which all the carbon–carbon bonds are single. Alkanes have the general chemical formula CnH2n+2. The alkanes range in complexity from the simplest case of methane, where n = 1, to arbitrarily large and complex molecules, like pentacontane or 6-ethyl-2-methyl-5-(1-methylethyl) octane, an isomer of tetradecane.

Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.
In chemistry, dehydrogenation is a chemical reaction that involves the removal of hydrogen, usually from an organic molecule. It is the reverse of hydrogenation. Dehydrogenation is important, both as a useful reaction and a serious problem. At its simplest, it's a useful way of converting alkanes, which are relatively inert and thus low-valued, to olefins, which are reactive and thus more valuable. Alkenes are precursors to aldehydes, alcohols, polymers, and aromatics. As a problematic reaction, the fouling and inactivation of many catalysts arises via coking, which is the dehydrogenative polymerization of organic substrates.
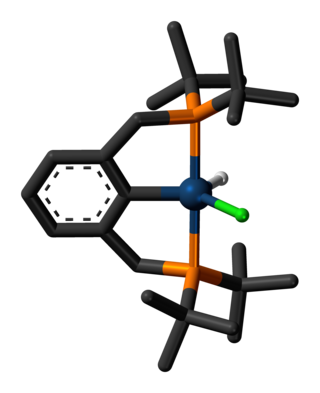
In chemistry, a transition metal pincer complex is a type of coordination complex with a pincer ligand. Pincer ligands are chelating agents that binds tightly to three adjacent coplanar sites in a meridional configuration. The inflexibility of the pincer-metal interaction confers high thermal stability to the resulting complexes. This stability is in part ascribed to the constrained geometry of the pincer, which inhibits cyclometallation of the organic substituents on the donor sites at each end. In the absence of this effect, cyclometallation is often a significant deactivation process for complexes, in particular limiting their ability to effect C-H bond activation. The organic substituents also define a hydrophobic pocket around the reactive coordination site. Stoichiometric and catalytic applications of pincer complexes have been studied at an accelerating pace since the mid-1970s. Most pincer ligands contain phosphines. Reactions of metal-pincer complexes are localized at three sites perpendicular to the plane of the pincer ligand, although in some cases one arm is hemi-labile and an additional coordination site is generated transiently. Early examples of pincer ligands were anionic with a carbanion as the central donor site and flanking phosphine donors; these compounds are referred to as PCP pincers.

Methyl bisulfate is a chemical compound with the molecular formula (CH3)HSO4. This compound is the mono-methyl ester of sulfuric acid. Its structure is CH3−O−S(=O)2−OH. The significance of methyl bisulfate is that it is an intermediate in the hydrolysis of the important reagent dimethyl sulfate, (CH3)2SO4:
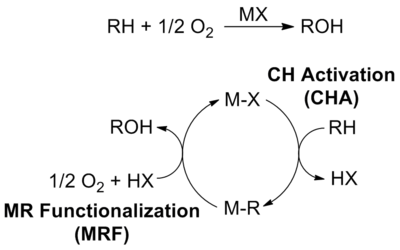
In organic chemistry and organometallic chemistry, carbon–hydrogen bond activation is a type of organic reaction in which a carbon–hydrogen bond is cleaved and replaced with a C−X bond. Some authors further restrict the term C–H activation to reactions in which a C–H bond, one that is typically considered to be "unreactive", interacts with a transition metal center M, resulting in its cleavage and the generation of an organometallic species with an M–C bond. The intermediate of this step could then undergo subsequent reactions with other reagents, either in situ or in a separate step, to produce the functionalized product.
In chemistry, carbonylation refers to reactions that introduce carbon monoxide (CO) into organic and inorganic substrates. Carbon monoxide is abundantly available and conveniently reactive, so it is widely used as a reactant in industrial chemistry. The term carbonylation also refers to oxidation of protein side chains.
In organometallic chemistry, a migratory insertion is a type of reaction wherein two ligands on a metal complex combine. It is a subset of reactions that very closely resembles the insertion reactions, and both are differentiated by the mechanism that leads to the resulting stereochemistry of the products. However, often the two are used interchangeably because the mechanism is sometimes unknown. Therefore, migratory insertion reactions or insertion reactions, for short, are defined not by the mechanism but by the overall regiochemistry wherein one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.:

The Shilov system is a classic example of catalytic C-H bond activation and oxidation which preferentially activates stronger C-H bonds over weaker C-H bonds for an overall partial oxidation.

Organoiridium chemistry is the chemistry of organometallic compounds containing an iridium-carbon chemical bond. Organoiridium compounds are relevant to many important processes including olefin hydrogenation and the industrial synthesis of acetic acid. They are also of great academic interest because of the diversity of the reactions and their relevance to the synthesis of fine chemicals.
Catalytic oxidation are processes that rely on catalysts to introduce oxygen into organic and inorganic compounds. Many applications, including the focus of this article, involve oxidation by oxygen. Such processes are conducted on a large scale for the remediation of pollutants, production of valuable chemicals, and the production of energy.
Organoplatinum chemistry is the chemistry of organometallic compounds containing a carbon to platinum chemical bond, and the study of platinum as a catalyst in organic reactions. Organoplatinum compounds exist in oxidation state 0 to IV, with oxidation state II most abundant. The general order in bond strength is Pt-C (sp) > Pt-O > Pt-N > Pt-C (sp3). Organoplatinum and organopalladium chemistry are similar, but organoplatinum compounds are more stable and therefore less useful as catalysts.

Organorhodium chemistry is the chemistry of organometallic compounds containing a rhodium-carbon chemical bond, and the study of rhodium and rhodium compounds as catalysts in organic reactions.
The oxidative coupling of methane (OCM) is a potential chemical reaction studied in the 1980s for the direct conversion of natural gas, primarily consisting of methane, into value-added chemicals. Although the reaction would have strong economics if practicable, no effective catalysts are known, and thermodynamic arguments suggest none can exist.
Georgiy Borisovich Shul’pin was born in Moscow, Russia. He graduated with a M.S. degree in chemistry from the Chemistry Department of Moscow State University in 1969. Between 1969 and 1972, he was a postgraduate student at the Nesmeyanov Institute of Organoelement Compounds under the direction of Prof. A. N. Nesmeyanov and received his Ph.D. in organometallic chemistry in 1975. He received his Dr. of Sciences degree in 2013.
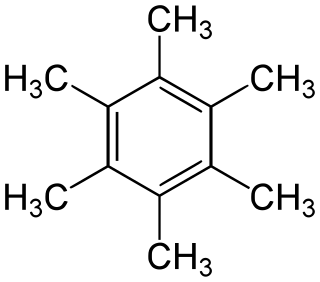
Hexamethylbenzene, also known as mellitene, is a hydrocarbon with the molecular formula C12H18 and the condensed structural formula C6(CH3)6. It is an aromatic compound and a derivative of benzene, where benzene's six hydrogen atoms have each been replaced by a methyl group. In 1929, Kathleen Lonsdale reported the crystal structure of hexamethylbenzene, demonstrating that the central ring is hexagonal and flat and thereby ending an ongoing debate about the physical parameters of the benzene system. This was a historically significant result, both for the field of X-ray crystallography and for understanding aromaticity.

The Scripps Energy & Materials Center (SEMC) is an American research center that focuses on research in the basic energy and materials sciences. Located in Jupiter, Florida, the center has scientists, graduate students, and administrative staff. The SEMC is a part of the Scripps Research Institute (TSRI), one of the largest non-profit research institutes in the world.
Methane functionalization is the process of converting methane in its gaseous state to another molecule with a functional group, typically methanol or acetic acid, through the use of transition metal catalysts.
Karen Ila Goldberg is an American chemist, currently the Vagelos Professor of Energy Research at University of Pennsylvania. Goldberg is most known for her work in inorganic and organometallic chemistry. Her most recent research focuses on catalysis, particularly on developing catalysts for oxidation, as well as the synthesis and activation of molecular oxygen. In 2018, Goldberg was elected to the National Academy of Sciences.

In organometallic chemistry, the activation of cyclopropanes by transition metals is a research theme with implications for organic synthesis and homogeneous catalysis. Being highly strained, cyclopropanes are prone to oxidative addition to transition metal complexes. The resulting metallacycles are susceptible to a variety of reactions. These reactions are rare examples of C-C bond activation. The rarity of C-C activation processes has been attributed to Steric effects that protect C-C bonds. Furthermore, the directionality of C-C bonds as compared to C-H bonds makes orbital interaction with transition metals less favorable. Thermodynamically, C-C bond activation is more favored than C-H bond activation as the strength of a typical C-C bond is around 90 kcal per mole while the strength of a typical unactivated C-H bond is around 104 kcal per mole.