
In bioinformatics, a sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. Sequence alignments are also used for non-biological sequences such as calculating the distance cost between strings in a natural language, or to display financial data.
Protein engineering is the process of developing useful or valuable proteins through the design and production of unnatural polypeptides, often by altering amino acid sequences found in nature. It is a young discipline, with much research taking place into the understanding of protein folding and recognition for protein design principles. It has been used to improve the function of many enzymes for industrial catalysis. It is also a product and services market, with an estimated value of $168 billion by 2017.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design.
In bioinformatics and evolutionary biology, a substitution matrix describes the frequency at which a character in a nucleotide sequence or a protein sequence changes to other character states over evolutionary time. The information is often in the form of log odds of finding two specific character states aligned and depends on the assumed number of evolutionary changes or sequence dissimilarity between compared sequences. It is an application of a stochastic matrix. Substitution matrices are usually seen in the context of amino acid or DNA sequence alignments, where they are used to calculate similarity scores between the aligned sequences.
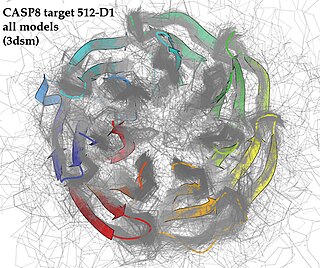
Critical Assessment of Structure Prediction (CASP), sometimes called Critical Assessment of Protein Structure Prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. CASP provides research groups with an opportunity to objectively test their structure prediction methods and delivers an independent assessment of the state of the art in protein structure modeling to the research community and software users. Even though the primary goal of CASP is to help advance the methods of identifying protein three-dimensional structure from its amino acid sequence many view the experiment more as a "world championship" in this field of science. More than 100 research groups from all over the world participate in CASP on a regular basis and it is not uncommon for entire groups to suspend their other research for months while they focus on getting their servers ready for the experiment and on performing the detailed predictions.

Structural alignment attempts to establish homology between two or more polymer structures based on their shape and three-dimensional conformation. This process is usually applied to protein tertiary structures but can also be used for large RNA molecules. In contrast to simple structural superposition, where at least some equivalent residues of the two structures are known, structural alignment requires no a priori knowledge of equivalent positions. Structural alignment is a valuable tool for the comparison of proteins with low sequence similarity, where evolutionary relationships between proteins cannot be easily detected by standard sequence alignment techniques. Structural alignment can therefore be used to imply evolutionary relationships between proteins that share very little common sequence. However, caution should be used in using the results as evidence for shared evolutionary ancestry because of the possible confounding effects of convergent evolution by which multiple unrelated amino acid sequences converge on a common tertiary structure.

Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
A Gap penalty is a method of scoring alignments of two or more sequences. When aligning sequences, introducing gaps in the sequences can allow an alignment algorithm to match more terms than a gap-less alignment can. However, minimizing gaps in an alignment is important to create a useful alignment. Too many gaps can cause an alignment to become meaningless. Gap penalties are used to adjust alignment scores based on the number and length of gaps. The five main types of gap penalties are constant, linear, affine, convex, and profile-based.
In molecular biology, protein threading, also known as fold recognition, is a method of protein modeling which is used to model those proteins which have the same fold as proteins of known structures, but do not have homologous proteins with known structure. It differs from the homology modeling method of structure prediction as it is used for proteins which do not have their homologous protein structures deposited in the Protein Data Bank (PDB), whereas homology modeling is used for those proteins which do. Threading works by using statistical knowledge of the relationship between the structures deposited in the PDB and the sequence of the protein which one wishes to model.

Multiple sequence alignment (MSA) is the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. These alignments are used to infer evolutionary relationships via phylogenetic analysis and can highlight homologous features between sequences. Alignments highlight mutation events such as point mutations, insertion mutations and deletion mutations, and alignments are used to assess sequence conservation and infer the presence and activity of protein domains, tertiary structures, secondary structures, and individual amino acids or nucleotides.
In bioinformatics, the root mean square deviation of atomic positions, or simply root mean square deviation (RMSD), is the measure of the average distance between the atoms (usually the backbone atoms) of superimposed molecules. In the study of globular protein conformations, one customarily measures the similarity in three-dimensional structure by the RMSD of the Cα atomic coordinates after optimal rigid body superposition.

Homology modeling, also known as comparative modeling of protein, refers to constructing an atomic-resolution model of the "target" protein from its amino acid sequence and an experimental three-dimensional structure of a related homologous protein. Homology modeling relies on the identification of one or more known protein structures likely to resemble the structure of the query sequence, and on the production of a sequence alignment that maps residues in the query sequence to residues in the template sequence. It has been seen that protein structures are more conserved than protein sequences amongst homologues, but sequences falling below a 20% sequence identity can have very different structure.
The global distance test (GDT), also written as GDT_TS to represent "total score", is a measure of similarity between two protein structures with known amino acid correspondences but different tertiary structures. It is most commonly used to compare the results of protein structure prediction to the experimentally determined structure as measured by X-ray crystallography, protein NMR, or, increasingly, cryoelectron microscopy.
The root mean square deviation (RMSD) or root mean square error (RMSE) is either one of two closely related and frequently used measures of the differences between true or predicted values on the one hand and observed values or an estimator on the other. The deviation is typically simply a differences of scalars; it can also be generalized to the vector lengths of a displacement, as in the bioinformatics concept of root mean square deviation of atomic positions.
CS-BLAST (Context-Specific BLAST) is a tool that searches a protein sequence that extends BLAST, using context-specific mutation probabilities. More specifically, CS-BLAST derives context-specific amino-acid similarities on each query sequence from short windows on the query sequences. Using CS-BLAST doubles sensitivity and significantly improves alignment quality without a loss of speed in comparison to BLAST. CSI-BLAST is the context-specific analog of PSI-BLAST, which computes the mutation profile with substitution probabilities and mixes it with the query profile. CSI-BLAST is the context specific analog of PSI-BLAST. Both of these programs are available as web-server and are available for free download.
The sequential structure alignment program (SSAP) in chemistry, physics, and biology is a method that uses double dynamic programming to produce a structural alignment based on atom-to-atom vectors in structure space. Instead of the alpha carbons typically used in structural alignment, SSAP constructs its vectors from the beta carbons for all residues except glycine, a method which thus takes into account the rotameric state of each residue as well as its location along the backbone. SSAP works by first constructing a series of inter-residue distance vectors between each residue and its nearest non-contiguous neighbors on each protein. A series of matrices are then constructed containing the vector differences between neighbors for each pair of residues for which vectors were constructed. Dynamic programming applied to each resulting matrix determines a series of optimal local alignments which are then summed into a "summary" matrix to which dynamic programming is applied again to determine the overall structural alignment.
I-sites are short sequence-structure motifs that are mined from the Protein Data Bank (PDB) that correlate strongly with three-dimensional structural elements. These sequence-structure motifs are used for the local structure prediction of proteins. Local structure can be expressed as fragments or as backbone angles. Locations in the protein sequence that have high confidence I-sites predictions may be the initiation sites of folding. I-sites have also been identified as discrete models for folding pathways. I-sites consist of about 250 motifs. Each motif has an amino acid profile, a fragment structure and optionally, a 4-dimensional tensor of pairwise sequence covariance.
RaptorX is a software and web server for protein structure and function prediction that is free for non-commercial use. RaptorX is among the most popular methods for protein structure prediction. Like other remote homology recognition and protein threading techniques, RaptorX is able to regularly generate reliable protein models when the widely used PSI-BLAST cannot. However, RaptorX is also significantly different from profile-based methods in that RaptorX excels at modeling of protein sequences without a large number of sequence homologs by exploiting structure information. RaptorX Server has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods.

I-TASSER is a bioinformatics method for predicting three-dimensional structure model of protein molecules from amino acid sequences. It detects structure templates from the Protein Data Bank by a technique called fold recognition. The full-length structure models are constructed by reassembling structural fragments from threading templates using replica exchange Monte Carlo simulations. I-TASSER is one of the most successful protein structure prediction methods in the community-wide CASP experiments.
SuperPose is a freely available web server designed to perform both pairwise and multiple protein structure superpositions. The “Structural superposition” term refers to the rotations and translations performed on one structure to make it match or align with another structure or structures. Structural superposition can be quantified either in terms of similarity or difference measures. The optimal superposition is the one in which the similarity measure is maximized or the difference measure is minimized. The “SuperPose” web server uses “RMSD” or Root-Mean-Square Deviation as a difference measure to find the optimal pairwise or multiple protein structure superposition. After an initial sequence and secondary structure alignment, SuperPose generates a Difference Distance (DD) matrix from the equivalent C-alpha atoms of two molecules. The sequence/structure alignment and DD matrix analysis information is then fed into a modified quaternion eigenvalue algorithm to rapidly perform the structural superposition and calculate the RMSD between aligned regions of two macromolecules.







