
Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

MASA syndrome is a rare X-linked recessive neurological disorder on the L1 disorder spectrum belonging in the group of hereditary spastic paraplegias a paraplegia known to increase stiffness spasticity in the lower limbs. This syndrome also has two other names, CRASH syndrome and Gareis-Mason syndrome.
Agenesis of the corpus callosum (ACC) is a rare birth defect in which there is a complete or partial absence of the corpus callosum. It occurs when the development of the corpus callosum, the band of white matter connecting the two hemispheres in the brain, in the embryo is disrupted. The result of this is that the fibers that would otherwise form the corpus callosum are instead longitudinally oriented along the ipsilateral ventricular wall and form structures called Probst bundles.

Zinc finger protein GLI3 is a protein that in humans is encoded by the GLI3 gene.

Greig cephalopolysyndactyly syndrome is a disorder that affects development of the limbs, head, and face. The features of this syndrome are highly variable, ranging from very mild to severe. People with this condition typically have one or more extra fingers or toes (polydactyly) or an abnormally wide thumb or big toe (hallux).

Pallister–Hall syndrome (PHS) is a rare genetic disorder that affects various body systems. The main features are a non-cancerous mass on the hypothalamus and extra digits (polydactylism). The prevalence of Pallister-Hall Syndrome is unknown; about 100 cases have been reported in publication.
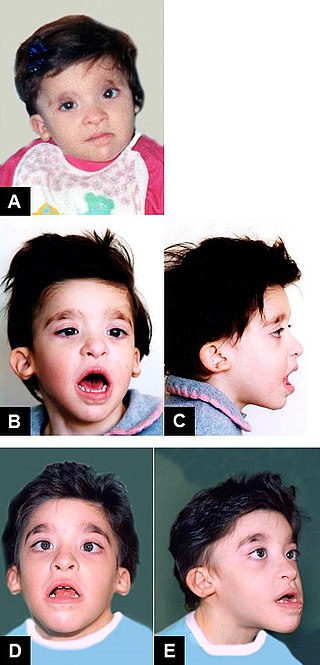
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998. The condition affects both males and females, has been described in various countries and ethnic groups around the world, and occurs in approximately 1 in 50,000–100,000 births.

3C syndrome is a rare condition whose symptoms include heart defects, cerebellar hypoplasia, and cranial dysmorphism. It was first described in the medical literature in 1987 by Ritscher and Schinzel, for whom the disorder is sometimes named.
Vici syndrome, also called immunodeficiency with cleft lip/palate, cataract, hypopigmentation and absent corpus callosum, is a rare autosomal recessive congenital disorder characterized by albinism, agenesis of the corpus callosum, cataracts, cardiomyopathy, severe psychomotor retardation, seizures, immunodeficiency and recurrent severe infections. To date, about 50 cases have been reported.

A ciliopathy is any genetic disorder that affects the cellular cilia or the cilia anchoring structures, the basal bodies, or ciliary function. Primary cilia are important in guiding the process of development, so abnormal ciliary function while an embryo is developing can lead to a set of malformations that can occur regardless of the particular genetic problem. The similarity of the clinical features of these developmental disorders means that they form a recognizable cluster of syndromes, loosely attributed to abnormal ciliary function and hence called ciliopathies. Regardless of the actual genetic cause, it is clustering of a set of characteristic physiological features which define whether a syndrome is a ciliopathy.

FG syndrome (FGS) is a rare genetic syndrome caused by one or more recessive genes located on the X chromosome and causing physical anomalies and developmental delays. FG syndrome was named after the first letters of the surnames of the first patients noted with the disease. First reported by American geneticists John M. Opitz and Elisabeth G. Kaveggia in 1974, its major clinical features include intellectual disability, hyperactivity, hypotonia, and a characteristic facial appearance including macrocephaly.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.

Andermann syndrome, also known as agenesis of corpus callosum with neuronopathy (ACCPN), Charlevoix disease and KCC3 axonopathy among other names, is a very rare neurodegenerative genetic disorder that damages the nerves used to control muscles and related to sensation and is often associated with agenesis of the corpus callosum.
Toriello–Carey syndrome is a genetic disorder that is characterized by Pierre Robin sequence and agenesis of the corpus callosum. Children with the disorder also possess a characteristic facial phenotype.
Chudley–Mccullough syndrome is a rare genetic disorder which is characterized by bilateral congenital hearing loss associated with brain malformations. It is a type of syndromic deafness.
Proud syndrome is a very rare genetic disorder which is characterized by severe intellectual disabilities, corpus callosum agenesis, epilepsy, and spasticity. It is a type of syndromic X-linked intellectual disability.
Holoprosencephaly-ectrodactyly-cleft lip/palate syndrome, also simply known as Hartsfield syndrome, is a rare genetic disorder characterized by the presence of variable holoprosencephaly, ectrodactyly, cleft lip and palate, alongside generalized ectodermal abnormalities. Additional findings include endocrine anomalies and developmental delays.
Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.

