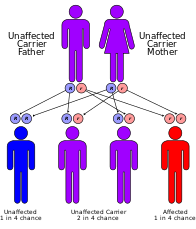
Werner syndrome (WS) or Werner's syndrome, also known as "adult progeria", is a rare, autosomal recessive disorder which is characterized by the appearance of premature aging.

Helicases are a class of enzymes thought to be vital to all organisms. Their main function is to unpack an organism's genetic material. Helicases are motor proteins that move directionally along a nucleic acid phosphodiester backbone, separating two hybridized nucleic acid strands, using energy from ATP hydrolysis. There are many helicases, representing the great variety of processes in which strand separation must be catalyzed. Approximately 1% of eukaryotic genes code for helicases.
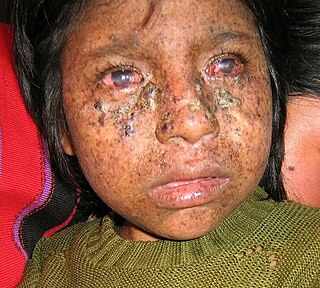
Xeroderma pigmentosum (XP) is a genetic disorder in which there is a decreased ability to repair DNA damage such as that caused by ultraviolet (UV) light. Symptoms may include a severe sunburn after only a few minutes in the sun, freckling in sun-exposed areas, dry skin and changes in skin pigmentation. Nervous system problems, such as hearing loss, poor coordination, loss of intellectual function and seizures, may also occur. Complications include a high risk of skin cancer, with about half having skin cancer by age 10 without preventative efforts, and cataracts. There may be a higher risk of other cancers such as brain cancers.

Nucleotide excision repair is a DNA repair mechanism. DNA damage occurs constantly because of chemicals, radiation and other mutagens. Three excision repair pathways exist to repair single stranded DNA damage: Nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR). While the BER pathway can recognize specific non-bulky lesions in DNA, it can correct only damaged bases that are removed by specific glycosylases. Similarly, the MMR pathway only targets mismatched Watson-Crick base pairs.
Chromosome instability syndromes are a group of inherited conditions associated with chromosomal instability and breakage. They often lead to an increased tendency to develop certain types of malignancies.

XPB is an ATP-dependent DNA helicase in humans that is a part of the TFIIH transcription factor complex.
A DNA repair-deficiency disorder is a medical condition due to reduced functionality of DNA repair.

ERCC2, or XPD is a protein involved in transcription-coupled nucleotide excision repair.

DeSanctis–Cacchione syndrome is a genetic disorder characterized by the skin and eye symptoms of xeroderma pigmentosum (XP) occurring in association with microcephaly, progressive intellectual disability, slowed growth and sexual development, deafness, choreoathetosis, ataxia and quadriparesis.

DNA excision repair protein ERCC-1 is a protein that in humans is encoded by the ERCC1 gene. Together with ERCC4, ERCC1 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.
The enzyme DNA-(apurinic or apyrimidinic site) lyase, also referred to as DNA-(apurinic or apyrimidinic site) 5'-phosphomonoester-lyase or DNA AP lyase catalyzes the cleavage of the C-O-P bond 3' from the apurinic or apyrimidinic site in DNA via β-elimination reaction, leaving a 3'-terminal unsaturated sugar and a product with a terminal 5'-phosphate. In the 1970s, this class of enzyme was found to repair at apurinic or apyrimidinic DNA sites in E. coli and in mammalian cells. The major active enzyme of this class in bacteria, and specifically, E. coli is endonuclease type III. This enzyme is part of a family of lyases that cleave carbon-oxygen bonds.

Xeroderma pigmentosum, complementation group C, also known as XPC, is a protein which in humans is encoded by the XPC gene. XPC is involved in the recognition of bulky DNA adducts in nucleotide excision repair. It is located on chromosome 3.

DNA repair protein complementing XP-A cells is a protein that in humans is encoded by the XPA gene.

DNA excision repair protein ERCC-6 is a protein that in humans is encoded by the ERCC6 gene. The ERCC6 gene is located on the long arm of chromosome 10 at position 11.23.

DNA repair protein complementing XP-G cells is a protein that in humans is encoded by the ERCC5 gene.
ERCC4 is a protein designated as DNA repair endonuclease XPF that in humans is encoded by the ERCC4 gene. Together with ERCC1, ERCC4 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.

DNA excision repair protein ERCC-8 is a protein that in humans is encoded by the ERCC8 gene.
Trichothiodystrophy (TTD) is an autosomal recessive inherited disorder characterised by brittle hair and intellectual impairment. The word breaks down into tricho – "hair", thio – "sulphur", and dystrophy – "wasting away" or literally "bad nourishment". TTD is associated with a range of symptoms connected with organs of the ectoderm and neuroectoderm. TTD may be subclassified into four syndromes: Approximately half of all patients with trichothiodystrophy have photosensitivity, which divides the classification into syndromes with or without photosensitivity; BIDS and PBIDS, and IBIDS and PIBIDS. Modern covering usage is TTD-P (photosensitive), and TTD.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.
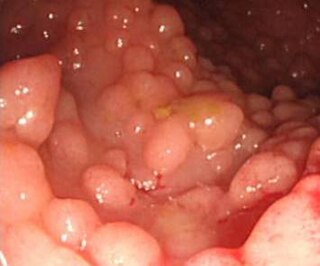
A hereditary cancer syndrome is a genetic disorder in which inherited genetic mutations in one or more genes predispose the affected individuals to the development of cancer and may also cause early onset of these cancers. Hereditary cancer syndromes often show not only a high lifetime risk of developing cancer, but also the development of multiple independent primary tumors.