
Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).

Neurofibromatosis (NF) refers to a group of three distinct genetic conditions in which tumors grow in the nervous system. The tumors are non-cancerous (benign) and often involve the skin or surrounding bone. Although symptoms are often mild, each condition presents differently. Neurofibromatosis type I (NF1) is typically characterized by café au lait spots, neurofibromas, scoliosis, and headaches. Neurofibromatosis type II (NF2), on the other hand, may present with early-onset hearing loss, cataracts, tinnitus, difficulty walking or maintain balance, and muscle atrophy. The third type is called schwannomatosis and often presents in early adulthood with widespread pain, numbness, or tingling due to nerve compression.
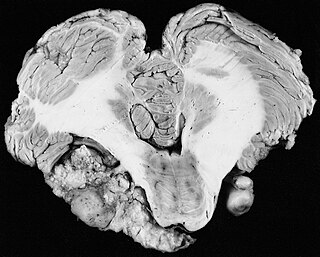
A vestibular schwannoma (VS), also called acoustic neuroma, is a benign tumor that develops on the vestibulocochlear nerve that passes from the inner ear to the brain. The tumor originates when Schwann cells that form the insulating myelin sheath on the nerve malfunction. Normally, Schwann cells function beneficially to protect the nerves which transmit balance and sound information to the brain. However, sometimes a mutation in the tumor suppressor gene, NF2, located on chromosome 22, results in abnormal production of the cell protein named Merlin, and Schwann cells multiply to form a tumor. The tumor originates mostly on the vestibular division of the nerve rather than the cochlear division, but hearing as well as balance will be affected as the tumor enlarges.
Spinal tumors are neoplasms located in either the vertebral column or the spinal cord. There are three main types of spinal tumors classified based on their location: extradural and intradural. Extradural tumors are located outside the dura mater lining and are most commonly metastatic. Intradural tumors are located inside the dura mater lining and are further subdivided into intramedullary and extramedullary tumors. Intradural-intramedullary tumors are located within the dura and spinal cord parenchyma, while intradural-extramedullary tumors are located within the dura but outside the spinal cord parenchyma. The most common presenting symptom of spinal tumors is nocturnal back pain. Other common symptoms include muscle weakness, sensory loss, and difficulty walking. Loss of bowel and bladder control may occur during the later stages of the disease.

Facial nerve paralysis is a common problem that involves the paralysis of any structures innervated by the facial nerve. The pathway of the facial nerve is long and relatively convoluted, so there are a number of causes that may result in facial nerve paralysis. The most common is Bell's palsy, a disease of unknown cause that may only be diagnosed by exclusion of identifiable serious causes.

A malignant peripheral nerve sheath tumor (MPNST) is a form of cancer of the connective tissue surrounding nerves. Given its origin and behavior it is classified as a sarcoma. About half the cases are diagnosed in people with neurofibromatosis; the lifetime risk for an MPNST in patients with neurofibromatosis type 1 is 8–13%. MPNST with rhabdomyoblastomatous component are called malignant triton tumors.

Neurofibromatosis type II is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves. The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s). Many people with this condition also experience vision problems. Neurofibromatosis type II is caused by mutations of the "Merlin" gene, which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.

A neurofibroma is a benign nerve-sheath tumor in the peripheral nervous system. In 90% of cases, they are found as stand-alone tumors, while the remainder are found in persons with neurofibromatosis type I (NF1), an autosomal-dominant genetically inherited disease. They can result in a range of symptoms from physical disfiguration and pain to cognitive disability.
Phakomatoses, also known as neurocutaneous syndromes, are a group of multisystemic diseases that most prominently affect structures primarily derived from the ectoderm such as the central nervous system, skin and eyes. The majority of phakomatoses are single-gene disorders that may be inherited in an autosomal dominant, autosomal recessive or X-linked pattern. Presentations may vary dramatically between patients with the same particular syndrome due to mosaicism, variable expressivity, and penetrance.
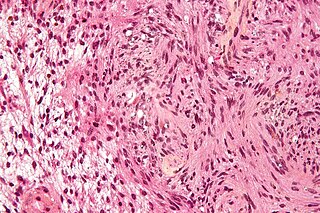
Schwannomatosis is an extremely rare genetic disorder closely related to the more-common disorder neurofibromatosis (NF). Originally described in Japanese patients, it consists of multiple cutaneous schwannomas, central nervous system tumors, and other neurological complications, excluding hallmark signs of NF. The exact frequency of schwannomatosis cases is unknown, although some populations have noted frequencies as few as 1 case per 1.7 million people.

A schwannoma is a usually benign nerve sheath tumor composed of Schwann cells, which normally produce the insulating myelin sheath covering peripheral nerves.

Neurofibromin 1 (NF1) is a gene in humans that is located on chromosome 17. NF1 codes for neurofibromin, a GTPase-activating protein that negatively regulates RAS/MAPK pathway activity by accelerating the hydrolysis of Ras-bound GTP. NF1 has a high mutation rate and mutations in NF1 can alter cellular growth control, and neural development, resulting in neurofibromatosis type 1. Symptoms of NF1 include disfiguring cutaneous neurofibromas (CNF), café au lait pigment spots, plexiform neurofibromas (PN), skeletal defects, optic nerve gliomas, life-threatening malignant peripheral nerve sheath tumors (MPNST), pheochromocytoma, attention deficits, learning deficits and other cognitive disabilities.
Juvenile myelomonocytic leukemia (JMML) is a rare form of chronic leukemia that affects children, commonly those aged four and younger. The name JMML now encompasses all diagnoses formerly referred to as juvenile chronic myeloid leukemia (JCML), chronic myelomonocytic leukemia of infancy, and infantile monosomy 7 syndrome. The average age of patients at diagnosis is two (2) years old. The World Health Organization has included JMML as a subcategory of myelodysplastic and myeloproliferative disorders.

Spinal disease refers to a condition impairing the backbone. These include various diseases of the back or spine ("dorso-"), such as kyphosis. Dorsalgia refers to back pain. Some other spinal diseases include spinal muscular atrophy, ankylosing spondylitis, scoliosis, lumbar spinal stenosis, spina bifida, spinal tumors, osteoporosis and cauda equina syndrome.

Bonnet–Dechaume–Blanc syndrome, also known as Wyburn-Mason syndrome, is a rare congenital disorder characterized by arteriovenous malformations of the brain, retina or facial nevi. The syndrome has a number of possible symptoms and can, more rarely, affect the skin, bones, kidneys, muscles, and gastrointestinal tract. When the syndrome affects the brain, people can experience severe headaches, seizures, acute stroke, meningism, and progressive neurological deficits due to acute or chronic ischaemia caused by arteriovenous shunting.

Legius syndrome (LS) is an autosomal dominant condition characterized by cafe au lait spots. It was first described in 2007 and is often mistaken for neurofibromatosis type I. It is caused by mutations in the SPRED1 gene. It is also known as neurofibromatosis type 1-like syndrome.

Selumetinib (INN), sold under the brand name Koselugo, is a medication for the treatment of children, two years of age and older, with neurofibromatosis type I (NF-1), a genetic disorder of the nervous system causing tumors to grow on nerves. It is taken by mouth.
Within medical ophthalmology, Intraocular schwannoma, also termed uveal schwannoma, is a type of schwannoma found in the eye. These tumors are almost always benign in nature and while malignant forms have been documented in other areas of the body, this has not been reported in the uveal region. Composed of Schwann cells, these masses are generally slow growing and can be found in the peripheral nerve tract, often around the head and neck.

Brigitte C. Widemann is German-American pediatric oncologist. She is chief of the pediatric oncology branch and clinical deputy director of the center for cancer research at the National Cancer Institute. She is also the special advisor to the NCI director for childhood cancer.
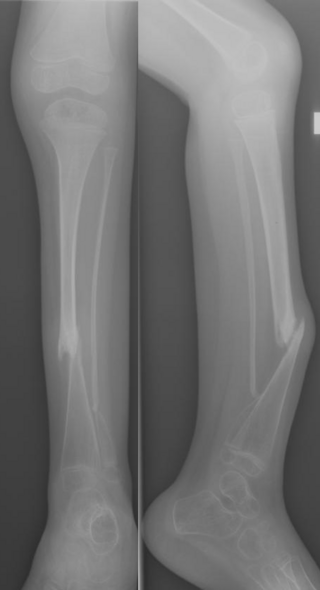
Congenital Pseudarthrosis of the Tibia (CPT) is a rare paediatric disease presenting with a bowing deformity of the tibia at birth or within the first decade of life. It is most commonly associated with Neurofibromatosis type 1 (NF-1). For children with CPT, pathological fracture of the tibia eventually occurs, resulting in persistent nonunion of the fracture site. If left untreated, leg deformities, joint stiffness, leg-length discrepancy and pain will persist. Diagnosis is done clinically and through X-ray imaging, with numerous classifications based on the severity of bowing and presence of fracture or intraosseous lesion.





{kind=link}