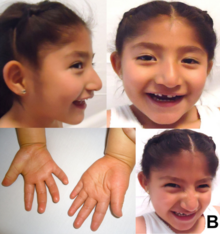
Prader–Willi syndrome (PWS) is a rare genetic disorder caused by a loss of function of specific genes on chromosome 15. In newborns, symptoms include weak muscles, poor feeding, and slow development. Beginning in childhood, those affected become constantly hungry, which often leads to obesity and type 2 diabetes. Mild to moderate intellectual impairment and behavioral problems are also typical of the disorder. Often, affected individuals have a narrow forehead, small hands and feet, short height, and light skin and hair. Most are unable to have children.

Chromosome 15q partial deletion is a rare human genetic disorder, caused by a chromosomal aberration in which the long ("q") arm of one copy of chromosome 15 is deleted, or partially deleted. Like other chromosomal disorders, this increases the risk of birth defects, developmental delay and learning difficulties, however, the problems that can develop depend very much on what genetic material is missing. If the mother's copy of the chromosomal region 15q11-13 is deleted, Angelman syndrome (AS) can result. The sister syndrome Prader-Willi syndrome (PWS) can result if the father's copy of the chromosomal region 15q11-13 is deleted. The smallest observed region that can result in these syndromes when deleted is therefore called the PWS/AS critical region. In addition to deletions, uniparental disomy of chromosome 15 also gives rise to the same genetic disorders, indicating that genomic imprinting must occur in this region.

In genetics, a deletion is a mutation in which a part of a chromosome or a sequence of DNA is left out during DNA replication. Any number of nucleotides can be deleted, from a single base to an entire piece of chromosome. Some chromosomes have fragile spots where breaks occur, which result in the deletion of a part of the chromosome. The breaks can be induced by heat, viruses, radiation, or chemical reactions. When a chromosome breaks, if a part of it is deleted or lost, the missing piece of chromosome is referred to as a deletion or a deficiency.
Jacobsen syndrome is a rare chromosomal disorder resulting from deletion of genes from chromosome 11 that includes band 11q24.1. It is a congenital disorder. Since the deletion takes place on the q arm of chromosome 11, it is also called 11q terminal deletion disorder. The deletion may range from 5 million to 16 million deleted DNA base pairs. The severity of symptoms depends on the number of deletions; the more deletions there are, the more severe the symptoms are likely to be.
Smith–Magenis syndrome (SMS), also known as 17p- syndrome, is a microdeletion syndrome characterized by an abnormality in the short (p) arm of chromosome 17. It has features including intellectual disability, facial abnormalities, difficulty sleeping, and numerous behavioral problems such as self-harm. Smith–Magenis syndrome affects an estimated between 1 in 15,000 to 1 in 25,000 individuals.

Ubiquitin-protein ligase E3A (UBE3A) also known as E6AP ubiquitin-protein ligase (E6AP) is an enzyme that in humans is encoded by the UBE3A gene. This enzyme is involved in targeting proteins for degradation within cells.
Chromosome 15 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 15 spans about 99.7 million base pairs and represents between 3% and 3.5% of the total DNA in cells. Chromosome 15 is an acrocentric chromosome, with a very small short arm, which contains few protein coding genes among its 19 million base pairs. It has a larger long arm that is gene rich, spanning about 83 million base pairs.
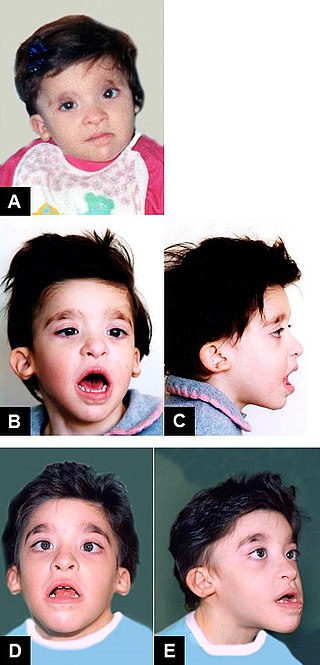
Mowat–Wilson syndrome is a rare genetic disorder that was clinically delineated by David R. Mowat and Meredith J. Wilson in 1998. The condition affects both males and females, has been described in various countries and ethnic groups around the world, and occurs in approximately 1 in 50,000–100,000 births.

22q13 deletion syndrome, known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

Isodicentric 15, also called marker chromosome 15 syndrome, idic(15), partial tetrasomy 15q, or inverted duplication 15, is a chromosome abnormality in which a child is born with extra genetic material from chromosome 15. People with idic(15) are typically born with 47 chromosomes in their body cells, instead of the normal 46. The extra chromosome, which is classified as a small supernumerary marker chromosome, is made up of a piece of chromosome 15 that has been duplicated end-to-end like a mirror image. It is the presence of this extra genetic material that is thought to account for the symptoms seen in some people with idic(15). Individuals with idic(15) have a total of four copies of this chromosome 15 region instead of the usual two copies. The term isodicentric refers to a duplication and inversion of a centromere-containing chromosomal segment.

Non-imprinted in Prader-Willi/Angelman syndrome region protein 1 is a protein that in humans is encoded by the NIPA1 gene. This gene encodes a potential transmembrane protein which functions either as a receptor or transporter molecule, possibly as a magnesium transporter. This protein is thought to play a role in nervous system development and maintenance. Alternative splice variants have been described, but their biological nature has not been determined. Mutations in this gene have been associated with the human genetic disease autosomal dominant spastic paraplegia 6.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, epilepsy, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Pitt–Hopkins syndrome can be marked by intellectual disabilities as well as problems with socializing. It is part of the clinical spectrum of Rett-like syndromes.

Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.

Sotos syndrome is a rare genetic disorder characterized by excessive physical growth during the first years of life. Excessive growth often starts in infancy and continues into the early teen years. The disorder may be accompanied by autism, mild intellectual disability, delayed motor, cognitive, and social development, hypotonia, and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have relatively large skulls (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disproportionately large skull with a slightly protrusive forehead, large hands and feet, large mandible, hypertelorism, and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur.

Distal 18q- is a genetic condition caused by a deletion of genetic material within one of the two copies of chromosome 18. The deletion involves the distal section of 18q and typically extends to the tip of the long arm of chromosome 18.

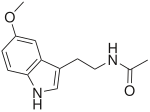
UBE3A-ATS/Ube3a-ATS (human/mouse), otherwise known as ubiquitin ligase E3A-ATS, is the name for the antisense DNA strand that is transcribed as part of a larger transcript called LNCAT at the Ube3a locus. The Ube3a locus is imprinted and in the central nervous system expressed only from the maternal allele. Silencing of Ube3a on the paternal allele is thought to occur through the Ube3a-ATS part of LNCAT, since non-coding antisense transcripts are often found at imprinted loci. The deletion and/or mutation of Ube3a on the maternal chromosome causes Angelman syndrome (AS) and Ube3a-ATS may prove to be an important aspect in finding a therapy for this disease. While in patients with AS the maternal Ube3a allele is inactive, the paternal allele is intact but epigenetically silenced. If unsilenced, the paternal allele could be a source of active Ube3a protein in AS patients. Therefore, understanding the mechanisms of how Ube3a-ATS might be involved in silencing the paternal Ube3a may lead to new therapies for AS. This possibility has been demonstrated by a recent study where the drug topotecan, administered to mice suffering from AS, activated expression of the paternal Ube3a gene by lowering the transcription of Ube3a-ATS.
Dup15q syndrome is the common name for maternally inherited chromosome 15q11.2-q13.1 duplication syndrome. This is a genomic copy number variant that leads to a type of neurodevelopmental disorder, caused by partial duplication of the proximal long arm of Chromosome 15. This variant confers a strong risk for autism spectrum disorder, epilepsy, and intellectual disability. It is the most common genetic cause of autism, accounting for approximately 1-3% of cases. Dup15q syndrome includes both interstitial duplications and isodicentric duplications of 15q11.2-13.1.
Chromosomal deletion syndromes result from deletion of parts of chromosomes. Depending on the location, size, and whom the deletion is inherited from, there are a few known different variations of chromosome deletions. Chromosomal deletion syndromes typically involve larger deletions that are visible using karyotyping techniques. Smaller deletions result in Microdeletion syndrome, which are detected using fluorescence in situ hybridization (FISH)

DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a syndrome caused by a microdeletion on the long arm of chromosome 22. While the symptoms can vary, they often include congenital heart problems, specific facial features, frequent infections, developmental disability, intellectual disability and cleft palate. Associated conditions include kidney problems, schizophrenia, hearing loss and autoimmune disorders such as rheumatoid arthritis or Graves' disease.
16p11.2 deletion syndrome is a rare genetic condition caused by microdeletion on the short arm of chromosome 16. Most affected individuals experience global developmental delay and intellectual disability, as well as childhood-onset obesity. 16p11.2 deletion is estimated to account for approximately 1% of autism spectrum disorder cases.