
An epileptic seizure, informally known as a seizure, is a period of symptoms due to abnormally excessive or synchronous neuronal activity in the brain. Outward effects vary from uncontrolled shaking movements involving much of the body with loss of consciousness, to shaking movements involving only part of the body with variable levels of consciousness, to a subtle momentary loss of awareness. Most of the time these episodes last less than two minutes and it takes some time to return to normal. Loss of bladder control may occur.
Absence seizures are one of several kinds of generalized seizures. In the past, absence epilepsy was referred to as "pyknolepsy," a term derived from the Greek word "pyknos," signifying "extremely frequent" or "grouped".These seizures are sometimes referred to as petit mal seizures ; however, usage of this terminology is no longer recommended

Tuberous sclerosis complex (TSC) is a rare multisystem autosomal dominant genetic disease that causes non-cancerous tumours to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, lung disease, and kidney disease.
Lennox–Gastaut syndrome (LGS) is a complex, rare, and severe childhood-onset epilepsy. It is characterized by multiple and concurrent seizure types including tonic seizure, cognitive dysfunction, and abnormal aspect on electroencephalogram (EEG) such as slow spike waves. Typically, it presents in children aged 3–5 years and most of the time persists into adulthood with slight changes in the electroclinical phentype. It has been associated with perinatal injuries, congenital infections, brain malformations, brain tumors, genetic disorders such as tuberous sclerosis and several gene mutations. Sometimes LGS is observed after infantile epileptic spasm syndrome. The prognosis for LGS is marked by a 5% mortality in childhood and persistent seizures into adulthood.
Frontal lobe epilepsy (FLE) is a neurological disorder that is characterized by brief, recurring seizures arising in the frontal lobes of the brain, that often occur during sleep. It is the second most common type of epilepsy after temporal lobe epilepsy (TLE), and is related to the temporal form in that both forms are characterized by partial (focal) seizures.
Dravet syndrome, previously known as severe myoclonic epilepsy of infancy (SMEI), Seizures are the most common form of DS. DS is diagnosed clinically and genetic testing is recommended if there is any doubt. Due to drug-refractory epilepsy in DS, many other therapies are being explored to prolong the life expectancy of patients.
A generalized tonic–clonic seizure, commonly known as a grand mal seizure or GTCS, is a type of generalized seizure that produces bilateral, convulsive tonic and clonic muscle contractions. Tonic–clonic seizures are the seizure type most commonly associated with epilepsy and seizures in general and the most common seizure associated with metabolic imbalances. It is a misconception that they are the sole type of seizure, as they are the main seizure type in approximately 10% of those with epilepsy.

GLUT1 deficiency syndrome, also known as GLUT1-DS, De Vivo disease or Glucose transporter type 1 deficiency syndrome, is an autosomal dominant genetic metabolic disorder associated with a deficiency of GLUT1, the protein that transports glucose across the blood brain barrier. Glucose Transporter Type 1 Deficiency Syndrome has an estimated birth incidence of 1 in 90,000 to 1 in 24,300. This birth incidence translates to an estimated prevalence of 3,000 to 7,000 in the U.S.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).
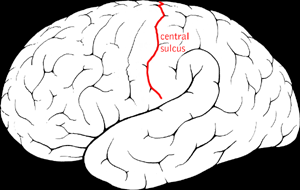
Benign Rolandic epilepsy or self-limited epilepsy with centrotemporal spikes is the most common epilepsy syndrome in childhood. Most children will outgrow the syndrome, hence the label benign. The seizures, sometimes referred to as sylvian seizures, start around the central sulcus of the brain.
Panayiotopoulos syndrome is a common idiopathic childhood-related seizure disorder that occurs exclusively in otherwise normal children and manifests mainly with autonomic epileptic seizures and autonomic status epilepticus. An expert consensus has defined Panayiotopoulos syndrome as "a benign age-related focal seizure disorder occurring in early and mid-childhood. It is characterized by seizures, often prolonged, with predominantly autonomic symptoms, and by an EEG [electroencephalogram] that shows shifting and/or multiple foci, often with occipital predominance."
Epilepsy is a neurological condition of recurrent episodes of unprovoked epileptic seizures. A seizure is an abnormal neuronal brain activity that can cause intellectual, emotional, and social consequences. Epilepsy affects children and adults of all ages and races. Epilepsy is one of the most common neurological disorders of the nervous system. Epilepsy is more common among children than adults affecting about 6 out of 1000 US children that are between the age of 0 to 5 years old. The epileptic seizures can be of different types depending on the part of the brain that was affected, seizures are classified in 2 main types partial seizure or genralized seizure.
Myoclonic astatic epilepsy (MAE), also known as myoclonic atonic epilepsy or Doose syndrome, is a generalized idiopathic epilepsy. It is characterized by the development of myoclonic seizures and/or myoclonic astatic seizures. Some of the common monogenic causes include mutations in the genes SLC6A1 (3p25.3),CHD2 (15q26.1), AP2M1 (10q23.2).
Idiopathic childhood occipital epilepsy of Gastaut (ICOE-G) is a pure but rare form of idiopathic occipital epilepsy that affects otherwise normal children and adolescents. It is classified amongst benign idiopathic childhood focal epilepsies such as rolandic epilepsy and Panayiotopoulos syndrome.
Jeavons syndrome is a type of epilepsy. It is one of the most distinctive reflex syndromes of idiopathic generalized epilepsy characterized by the triad of eyelid myoclonia with and without absences, eye-closure-induced seizures, EEG paroxysms, or both, and photosensitivity. Eyelid myoclonia with or without absences is a form of epileptic seizure manifesting with myoclonic jerks of the eyelids with or without a brief absence. These are mainly precipitated by closing of the eyes and lights. Eyelid myoclonia is the defining seizure type of Jeavons syndrome.
Epilepsy-intellectual disability in females also known as PCDH19 gene-related epilepsy or epileptic encephalopathy, early infantile, 9 (EIEE9), is a rare type of epilepsy that affects predominately females and is characterized by clusters of brief seizures, which start in infancy or early childhood, and is occasionally accompanied by varying degrees of cognitive impairment. The striking pattern of onset seizures at a young age, genetic testing and laboratory results, potential developmental delays or developmental regression and associated disorders, eases diagnosis.
Febrile infection-related epilepsy syndrome (FIRES), is onset of severe seizures following a febrile illness in someone who was previously healthy. The seizures may initially be focal; however, often become tonic-clonic. Complications often include intellectual disability, behavioral problems, and ongoing seizures.
People with epilepsy may be classified into different syndromes based on specific clinical features. These features include the age at which seizures begin, the seizure types, and EEG findings, among others. Identifying an epilepsy syndrome is useful as it helps determine the underlying causes as well as deciding what anti-seizure medication should be tried. Epilepsy syndromes are more commonly diagnosed in infants and children. Some examples of epilepsy syndromes include benign rolandic epilepsy, childhood absence epilepsy and juvenile myoclonic epilepsy. Severe syndromes with diffuse brain dysfunction caused, at least partly, by some aspect of epilepsy, are also referred to as epileptic encephalopathies. These are associated with frequent seizures that are resistant to treatment and severe cognitive dysfunction, for instance Lennox-Gastaut syndrome and West syndrome.
Drug-resistant epilepsy (DRE), also known as refractory epilepsy, intractable epilepsy, or pharmacoresistant epilepsy, is diagnosed following a failure of adequate trials of two tolerated and appropriately chosen and used antiepileptic drugs (AEDs) to achieve sustained seizure freedom. The probability that the next medication will achieve seizure freedom drops with every failed AED. For example, after two failed AEDs, the probability that the third will achieve seizure freedom is around 4%. Drug-resistant epilepsy is commonly diagnosed after several years of uncontrolled seizures, however, in most cases, it is evident much earlier. Approximately 30% of people with epilepsy have a drug-resistant form.
Malignant migrating partial seizures of infancy (MMPSI) is a rare epileptic syndrome that onsets before 6 months of age, commonly in the first few weeks of life. Once seizures start, the site of seizure activity repeatedly migrates from one area of the brain to another, with few periods of remission in between. These seizures are 'focal' (updated term for 'partial'), meaning they do not affect both sides of the brain at the same time. These continuous seizures cause damage to the brain, hence the descriptor 'malignant.'



