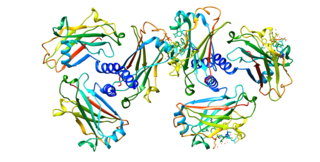
CD30, also known as TNFRSF8, is a cell membrane protein of the tumor necrosis factor receptor family and a tumor marker.
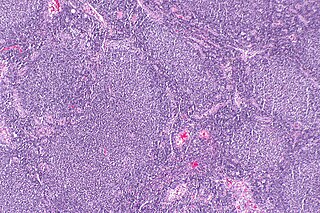
Follicular lymphoma (FL) is a cancer that involves certain types of white blood cells known as lymphocytes. The cancer originates from the uncontrolled division of specific types of B-cells known as centrocytes and centroblasts. These cells normally occupy the follicles in the germinal centers of lymphoid tissues such as lymph nodes. The cancerous cells in FL typically form follicular or follicle-like structures in the tissues they invade. These structures are usually the dominant histological feature of this cancer.
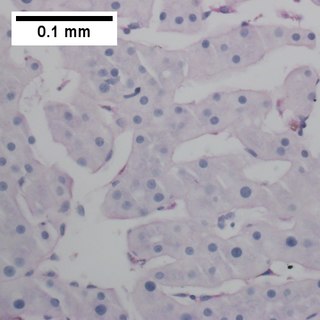
Primary effusion lymphoma (PEL) is classified as a diffuse large B cell lymphoma. It is a rare malignancy of plasmablastic cells that occurs in individuals that are infected with the Kaposi's sarcoma-associated herpesvirus. Plasmablasts are immature plasma cells, i.e. lymphocytes of the B-cell type that have differentiated into plasmablasts but because of their malignant nature do not differentiate into mature plasma cells but rather proliferate excessively and thereby cause life-threatening disease. In PEL, the proliferating plasmablastoid cells commonly accumulate within body cavities to produce effusions, primarily in the pleural, pericardial, or peritoneal cavities, without forming a contiguous tumor mass. In rare cases of these cavitary forms of PEL, the effusions develop in joints, the epidural space surrounding the brain and spinal cord, and underneath the capsule which forms around breast implants. Less frequently, individuals present with extracavitary primary effusion lymphomas, i.e., solid tumor masses not accompanied by effusions. The extracavitary tumors may develop in lymph nodes, bone, bone marrow, the gastrointestinal tract, skin, spleen, liver, lungs, central nervous system, testes, paranasal sinuses, muscle, and, rarely, inside the vasculature and sinuses of lymph nodes. As their disease progresses, however, individuals with the classical effusion-form of PEL may develop extracavitary tumors and individuals with extracavitary PEL may develop cavitary effusions.

T-cell lymphoma is a rare form of cancerous lymphoma affecting T-cells. Lymphoma arises mainly from the uncontrolled proliferation of T-cells and can become cancerous.

Intravascular lymphomas (IVL) are rare cancers in which malignant lymphocytes proliferate and accumulate within blood vessels. Almost all other types of lymphoma involve the proliferation and accumulation of malignant lymphocytes in lymph nodes, other parts of the lymphatic system, and various non-lymphatic organs but not in blood vessels.
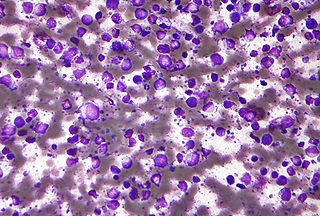
Diffuse large B-cell lymphoma (DLBCL) is a cancer of B cells, a type of lymphocyte that is responsible for producing antibodies. It is the most common form of non-Hodgkin lymphoma among adults, with an annual incidence of 7–8 cases per 100,000 people per year in the US and UK. This cancer occurs primarily in older individuals, with a median age of diagnosis at ~70 years, although it can occur in young adults and, in rare cases, children. DLBCL can arise in virtually any part of the body and, depending on various factors, is often a very aggressive malignancy. The first sign of this illness is typically the observation of a rapidly growing mass or tissue infiltration that is sometimes associated with systemic B symptoms, e.g. fever, weight loss, and night sweats.

Anaplastic lymphoma kinase (ALK) also known as ALK tyrosine kinase receptor or CD246 is an enzyme that in humans is encoded by the ALK gene.
Enteropathy-associated T-cell lymphoma (EATL), previously termed enteropathy-associated T-cell lymphoma, type I and at one time termed enteropathy-type T-cell lymphoma (ETTL), is a complication of coeliac disease in which a malignant T-cell lymphoma develops in areas of the small intestine affected by the disease's intense inflammation. While a relatively rare disease, it is the most common type of primary gastrointestinal T-cell lymphoma.

Marginal zone lymphomas, also known as marginal zone B-cell lymphomas (MZLs), are a heterogeneous group of lymphomas that derive from the malignant transformation of marginal zone B-cells. Marginal zone B cells are innate lymphoid cells that normally function by rapidly mounting IgM antibody immune responses to antigens such as those presented by infectious agents and damaged tissues. They are lymphocytes of the B-cell line that originate and mature in secondary lymphoid follicles and then move to the marginal zones of mucosa-associated lymphoid tissue (MALT), the spleen, or lymph nodes. Mucosa-associated lymphoid tissue is a diffuse system of small concentrations of lymphoid tissue found in various submucosal membrane sites of the body such as the gastrointestinal tract, mouth, nasal cavity, pharynx, thyroid gland, breast, lung, salivary glands, eye, skin and the human spleen.

Extranodal NK/T-cell lymphoma, nasal type (ENKTCL-NT) is a rare type of lymphoma that commonly involves midline areas of the nasal cavity, oral cavity, and/or pharynx At these sites, the disease often takes the form of massive, necrotic, and extremely disfiguring lesions. However, ENKTCL-NT can also involve the eye, larynx, lung, gastrointestinal tract, skin, and various other tissues. ENKTCL-NT mainly affects adults; it is relatively common in Asia and to lesser extents Mexico, Central America, and South America but is rare in Europe and North America. In Korea, ENKTCL-NT often involves the skin and is reported to be the most common form of cutaneous lymphoma after mycosis fungoides.
Brentuximab vedotin, sold under the brand name Adcetris, is an antibody-drug conjugate medication used to treat relapsed or refractory Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (ALCL), a type of T cell non-Hodgkin lymphoma. It selectively targets tumor cells expressing the CD30 antigen, a defining marker of Hodgkin lymphoma and ALCL. The drug is being jointly marketed by Millennium Pharmaceuticals outside the US and by Seagen in the US.

Crizotinib, sold under the brand name Xalkori among others, is an anti-cancer medication used for the treatment of non-small cell lung carcinoma (NSCLC). It acts as an ALK and ROS1 inhibitor.

Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm of the mesodermal cells that form the connective tissues which support virtually all of the organs and tissues of the body. IMT was formerly termed inflammatory pseudotumor. Currently, however, inflammatory pseudotumor designates a large and heterogeneous group of soft tissue tumors that includes inflammatory myofibroblastic tumor, plasma cell granuloma, xanthomatous pseudotumor, solitary mast cell granuloma, inflammatory fibrosarcoma, pseudosarcomatous myofibroblastic proliferation, myofibroblastoma, inflammatory myofibrohistiocytic proliferation, and other tumors that develop from connective tissue cells. Inflammatory pseudotumour is a generic term applied to various neoplastic and non-neoplastic tissue lesions which share a common microscopic appearance consisting of spindle cells and a prominent presence of the white blood cells that populate chronic or, less commonly, acute inflamed tissues.

Plasmablastic lymphoma (PBL) is a type of large B-cell lymphoma recognized by the World Health Organization (WHO) in 2017 as belonging to a subgroup of lymphomas termed lymphoid neoplasms with plasmablastic differentiation. The other lymphoid neoplasms within this subgroup are: plasmablastic plasma cell lymphoma ; primary effusion lymphoma that is Kaposi's sarcoma-associated herpesvirus positive or Kaposi's sarcoma-associated Herpesvirus negative; anaplastic lymphoma kinase-positive large B-cell lymphoma; and human herpesvirus 8-positive diffuse large B-cell lymphoma, not otherwise specified. All of these lymphomas are malignancies of plasmablasts, i.e. B-cells that have differentiated into plasmablasts but because of their malignant nature: fail to differentiate further into mature plasma cells; proliferate excessively; and accumulate in and injure various tissues and organs.
Seagen Inc. is an American biotechnology company focused on developing and commercializing innovative, empowered monoclonal antibody-based therapies for the treatment of cancer. The company, headquartered in Bothell, Washington, is the industry leader in antibody-drug conjugates or ADCs, a technology designed to harness the targeting ability of monoclonal antibodies to deliver cell-killing agents directly to cancer cells. Antibody-drug conjugates are intended to spare non-targeted cells and thus reduce many of the toxic effects of traditional chemotherapy, while potentially enhancing antitumor activity.
ALK+ large B-cell lymphoma is a type of lymphoma. It was first reported in 1997. It is a rare, aggressive large B-cell process that shows ALK expression. It is distinct from anaplastic large cell lymphoma, a T-cell lymphoma.
Monomorphic epitheliotropic intestinal T cell lymphoma (MEITL) is an extremely rare peripheral T-cell lymphoma that involves the malignant proliferation of a type of lymphocyte, the T cell, in the gastrointestinal tract. Over time, these T cells commonly spread throughout the mucosal lining of a portion of the GI tract, lead to GI tract nodules and ulcerations, and cause symptoms such as abdominal pain, weight loss, diarrhea, obstruction, bleeding, and/or perforation.
Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL-LT) is a cutaneous lymphoma skin disease that occurs mostly in elderly females. In this disease, B cells become malignant, accumulate in the dermis and subcutaneous tissue below the dermis to form red and violaceous skin nodules and tumors. These lesions typically occur on the lower extremities but in uncommon cases may develop on the skin at virtually any other site. In ~10% of cases, the disease presents with one or more skin lesions none of which are on the lower extremities; the disease in these cases is sometimes regarded as a variant of PCDLBL, LT termed primary cutaneous diffuse large B-cell lymphoma, other (PCDLBC-O). PCDLBCL, LT is a subtype of the diffuse large B-cell lymphomas (DLBCL) and has been thought of as a cutaneous counterpart to them. Like most variants and subtypes of the DLBCL, PCDLBCL, LT is an aggressive malignancy. It has a 5-year overall survival rate of 40–55%, although the PCDLBCL-O variant has a better prognosis than cases in which the legs are involved.
Diffuse large B-cell lymphoma associated with chronic inflammation (DLBCL-CI) is a subtype of the Diffuse large B-cell lymphomas and a rare form of the Epstein–Barr virus-associated lymphoproliferative diseases, i.e. conditions in which lymphocytes infected with the Epstein-Barr virus (EBV) proliferate excessively in one or more tissues. EBV infects ~95% of the world's population to cause no symptoms, minor non-specific symptoms, or infectious mononucleosis. The virus then enters a latency phase in which the infected individual becomes a lifetime asymptomatic carrier of the virus. Some weeks, months, years, or decades thereafter, a very small fraction of these carriers, particularly those with an immunodeficiency, develop any one of various EBV-associated benign or malignant diseases.
Mature T-cell lymphoma, also called peripheral T-cell lymphoma, is a group of rare, aggressive lymphomas that develop from mature white blood cells and originate from lymphoid tissues outside of the bone marrow. Mature T-cell lymphoma is under the category of non-Hodgkin lymphoma. Mature T-cell lymphomas account for 10% to 15% of all lymphomas and is more common in Asia than in Europe and America. Its common subtypes include angioimmunoblastic T-cell lymphoma, anaplastic large cell lymphoma and peripheral T-cell lymphoma not otherwise specified. While different subtypes have variable symptoms, common symptoms include enlarged painless lymph nodes, fever, weight loss, rash and night sweats.

