
5α-Reductase 2 deficiency (5αR2D) is an autosomal recessive condition caused by a mutation in SRD5A2, a gene encoding the enzyme 5α-reductase type 2 (5αR2). The condition is rare, affects only genetic males, and has a broad spectrum.
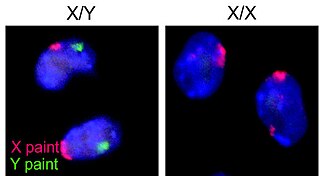
The XY sex-determination system is a sex-determination system used to classify many mammals, including humans, some insects (Drosophila), some snakes, some fish (guppies), and some plants. In this system, the sex of an individual is determined by a pair of sex chromosomes. Females have two of the same kind of sex chromosome (XX), and are called the homogametic sex. Males have two different kinds of sex chromosomes (XY), and are called the heterogametic sex.

Androgen insensitivity syndrome (AIS) is a condition involving the inability to respond to androgens, typically due to androgen receptor dysfunction.

XY complete gonadal dysgenesis, also known as Swyer syndrome, is a type of defect hypogonadism in a person whose karyotype is 46,XY. Though they typically have normal vulvas, the person has underdeveloped gonads, fibrous tissue termed "streak gonads", and if left untreated, will not experience puberty. The cause is a lack or inactivation of an SRY gene which is responsible for sexual differentiation. Pregnancy is often possible in Swyer syndrome with assisted reproductive technology. The phenotype is usually similar to Turner syndrome (45,X0) due to a lack of X inactivation. The typical medical treatment is hormone replacement therapy. The syndrome was named after Gerald Swyer, an endocrinologist based in London.
Virilization or masculinization is the biological development of adult male characteristics in young males or females. Most of the changes of virilization are produced by androgens.

Sex-determining region Y protein (SRY), or testis-determining factor (TDF), is a DNA-binding protein encoded by the SRY gene that is responsible for the initiation of male sex determination in therian mammals. SRY is an intronless sex-determining gene on the Y chromosome. Mutations in this gene lead to a range of disorders of sex development with varying effects on an individual's phenotype and genotype.

Ovotesticular syndrome is a rare congenital condition where an individual is born with both ovarian and testicular tissue. It is one of the rarest DSDs, with only 500 reported cases. Commonly, one or both gonads is an ovotestis containing both types of tissue. Although it is similar in some ways to mixed gonadal dysgenesis, the conditions can be distinguished histologically.
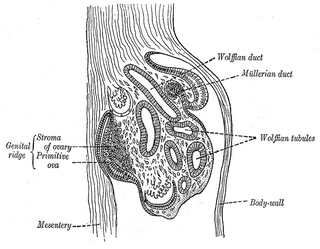
The genital ridge is the precursor to the gonads. The genital ridge initially consists mainly of mesenchyme and cells of underlying mesonephric origin. Once oogonia enter this area they attempt to associate with these somatic cells. Development proceeds and the oogonia become fully surrounded by a layer of cells.

XXYY syndrome is a sex chromosome anomaly in which males have 2 extra chromosomes, one X and one Y chromosome. Human cells usually contain two sex chromosomes, one from the mother and one from the father. Usually, females have two X chromosomes (XX) and males have one X and one Y chromosome (XY). The appearance of at least one Y chromosome with a properly functioning SRY gene makes a male. Therefore, humans with XXYY are genotypically male. Males with XXYY syndrome have 48 chromosomes instead of the typical 46. This is why XXYY syndrome is sometimes written as 48, XXYY syndrome or 48, XXYY. It affects an estimated one in every 18,000–40,000 male births.

Gonadal dysgenesis is classified as any congenital developmental disorder of the reproductive system in humans. It is atypical development of gonads in an embryo. One type of gonadal dysgenesis is the development of functionless, fibrous tissue, termed streak gonads, instead of reproductive tissue. Streak gonads are a form of aplasia, resulting in hormonal failure that manifests as sexual infantism and infertility, with no initiation of puberty and secondary sex characteristics.
Sex chromosomes are chromosomes that carry the genes that determine the sex of an individual. The human sex chromosomes are a typical pair of mammal allosomes. They differ from autosomes in form, size, and behavior. Whereas autosomes occur in homologous pairs whose members have the same form in a diploid cell, members of an allosome pair may differ from one another.

Sexual differentiation in humans is the process of development of sex differences in humans. It is defined as the development of phenotypic structures consequent to the action of hormones produced following gonadal determination. Sexual differentiation includes development of different genitalia and the internal genital tracts and body hair plays a role in sex identification.
Disorders of sex development (DSDs), also known as differences in sex development or variations in sex characteristics (VSC), are congenital conditions affecting the reproductive system, in which development of chromosomal, gonadal, or anatomical sex is atypical.

The steroidogenic factor 1 (SF-1) protein is a transcription factor involved in sex determination by controlling the activity of genes related to the reproductive glands or gonads and adrenal glands. This protein is encoded by the NR5A1 gene, a member of the nuclear receptor subfamily, located on the long arm of chromosome 9 at position 33.3. It was originally identified as a regulator of genes encoding cytochrome P450 steroid hydroxylases, however, further roles in endocrine function have since been discovered.
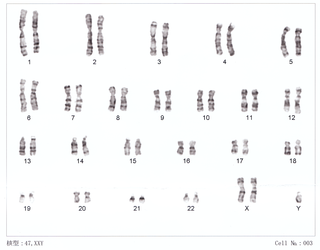
Klinefelter syndrome (KS), also known as 47,XXY, is a chromosome anomaly where a male has an extra X chromosome. These complications commonly include infertility and small, poorly functioning testicles. These symptoms are often noticed only at puberty, although this is one of the most common chromosomal disorders, occurring in one to two per 1,000 live births. It is named after American endocrinologist Harry Klinefelter, who identified the condition in the 1940s.
46,XX/46,XY is a chimeric genetic condition characterized by the presence of some cells that express a 46,XX karyotype and some cells that express a 46,XY karyotype in a single human being. The cause of the condition lies in utero with the aggregation of two distinct blastocysts or zygotes into a single embryo, which subsequently leads to the development of a single individual with two distinct cell lines, instead of a pair of fraternal twins. 46,XX/46,XY chimeras are the result of the merging of two non-identical twins. This is not to be confused with mosaicism or hybridism, neither of which are chimeric conditions.
45,X/46,XY mosaicism, also known as X0/XY mosaicism and mixed gonadal dysgenesis, is a mutation of sex development in humans associated with sex chromosome aneuploidy and mosaicism of the Y chromosome. It is a fairly rare chromosomal disorder at birth, with an estimated incidence rate of about 1 in 15,000 live births. Mosaic loss of the Y chromosome in previously non-mosaic men grows increasingly common with age.
Albert Fredrik de la Chapelle, MD, Ph.D was a Finnish human geneticist, long-time head of Finland's first Department of Medical Genetics at the University of Helsinki, and subsequently professor of Human Cancer Genetics at Ohio State University. He was best known for his role in the elucidation of the genetics of hereditary colorectal cancer and Lynch syndrome.
Sexual anomalies, also known as sexual abnormalities, are a set of clinical conditions due to chromosomal, gonadal and/or genitalia variation. Individuals with congenital (inborn) discrepancy between sex chromosome, gonadal, and their internal and external genitalia are categorised as individuals with a disorder of sex development (DSD). Afterwards, if the family or individual wishes, they can partake in different management and treatment options for their conditions.
Various criteria have been offered for the definition of intersex, including ambiguous genitalia, atypical genitalia, and differential sexual development. Ambiguous genitalia occurs in roughly 0.05% of all births, and atypical genitalia occurs in 0.5% of all births, usually caused by masculinization or feminization during pregnancy, these conditions range from full androgen insensitivity syndrome to ovotesticular syndrome, although the definition of what constitutes "normal" genitalia is largely arbitrary.




