
A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Von Willebrand disease (VWD) is the most common hereditary blood-clotting disorder in humans. An acquired form can sometimes result from other medical conditions. It arises from a deficiency in the quality or quantity of von Willebrand factor (VWF), a multimeric protein that is required for platelet adhesion. It is known to affect several breeds of dogs as well as humans. The three forms of VWD are hereditary, acquired, and pseudo or platelet type. The three types of hereditary VWD are VWD type 1, VWD type 2, and VWD type 3. Type 2 contains various subtypes. Platelet type VWD is also an inherited condition.
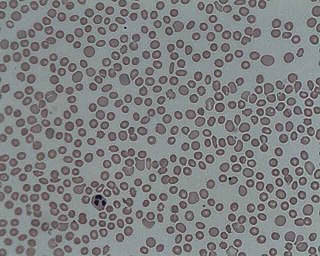
In hematology, thrombocytopenia is a condition characterized by abnormally low levels of platelets in the blood. Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.
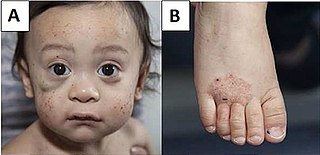
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea. It is also sometimes called the eczema-thrombocytopenia-immunodeficiency syndrome in keeping with Aldrich's original description in 1954. The WAS-related disorders of X-linked thrombocytopenia (XLT) and X-linked congenital neutropenia (XLN) may present with similar but less severe symptoms and are caused by mutations of the same gene.
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections. It causes severe pyogenic infections. It can be caused by autosomal dominant inheritance of the ELANE gene, autosomal recessive inheritance of the HAX1 gene. There is an increased risk of leukemia and myelodysplastic cancers.

Heřmanský–Pudlák syndrome is an extremely rare autosomal recessive disorder which results in oculocutaneous albinism, bleeding problems due to a platelet abnormality, and storage of an abnormal fat-protein compound. It is thought to affect around 1 in 500,000 people worldwide, with a significantly higher occurrence in Puerto Ricans, with a prevalence of 1 in 1800. Many of the clinical research studies on the disease have been conducted in Puerto Rico.
Mean platelet volume (MPV) is a machine-calculated measurement of the average size of platelets found in blood and is typically included in blood tests as part of the CBC. Since the average platelet size is larger when the body is producing increased numbers of platelets, the MPV test results can be used to make inferences about platelet production in bone marrow or platelet destruction problems.

Duane-radial ray syndrome, also known as Okihiro Syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.
Gray platelet syndrome (GPS), or platelet alpha-granule deficiency, is a rare congenital autosomal recessive bleeding disorder caused by a reduction or absence of alpha-granules in blood platelets, and the release of proteins normally contained in these granules into the marrow, causing myelofibrosis. The name derives from the initial observation of gray appearance of platelets with a paucity of granules on blood films from a patient with a lifelong bleeding disorder.

May–Hegglin anomaly (MHA), is a rare genetic disorder of the blood platelets that causes them to be abnormally large.
Pearson syndrome is a mitochondrial disease characterized by sideroblastic anemia and exocrine pancreas dysfunction. Other clinical features are failure to thrive, pancreatic fibrosis with insulin-dependent diabetes and exocrine pancreatic deficiency, muscle and neurologic impairment, and, frequently, early death. It is usually fatal in infancy. The few patients who survive into adulthood often develop symptoms of Kearns–Sayre syndrome. It is caused by a deletion in mitochondrial DNA. Pearson syndrome is very rare, less than a hundred cases have been reported in medical literature worldwide.
Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare autosomal recessive bone marrow failure syndrome characterized by severe thrombocytopenia, which can progress to aplastic anemia and leukemia. CAMT usually manifests as thrombocytopenia in the initial month of life or in the fetal phase. Typically CAMPT presents with petechiae, cerebral bleeds, recurrent rectal bleeding, or pulmonary hemorrhage.
Lix1 homolog (mouse)-like also known as LIX1L is a protein which in humans is encoded by the LIX1L gene.
Ankyrin repeat domain 35 also known as ANKRD35 is a protein which in humans is encoded by the ANKRD35 gene.
Polymerase (RNA) III polypeptide G (32kD)-like also known as POLR3GL is a protein which in humans is encoded by the POLR3GL gene.
Harris platelet syndrome (HPS) is the most common inherited giant platelet disorder.
1q21.1 deletion syndrome is a rare aberration of chromosome 1. A human cell has one pair of identical chromosomes on chromosome 1. With the 1q21.1 deletion syndrome, one chromosome of the pair is not complete, because a part of the sequence of the chromosome is missing. One chromosome has the normal length and the other is too short.
1q21.1 duplication syndrome or 1q21.1 (recurrent) microduplication is a rare aberration of chromosome 1.

Epstein syndrome is a rare genetic disease characterized by a mutation in the MYH9 gene in nonmuscle myosin. This disease affects the patient's renal system and can result in kidney failure. Epstein Syndrome was first discovered in 1972 when two families had similar symptoms to Alport syndrome. Epstein syndrome and other Alport-like disorders were seen to be caused by mutations in the MYH9 gene, however, Epstein syndrome differs as it was more specifically linked to a mutation on the R702 codon on the MYH9 gene. Diseases with mutations on the MYH9 gene also include May–Hegglin anomaly, Sebastian syndrome and Fechtner syndrome.
Toriello–Carey syndrome is a genetic disorder that is characterized by Pierre Robin sequence and agenesis of the corpus callosum. Children with the disorder also possess a characteristic facial phenotype.
