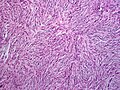
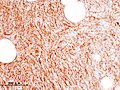
A sarcoma is a malignant tumor, a type of cancer that arises from cells of mesenchymal origin. Connective tissue is a broad term that includes bone, cartilage, fat, vascular, or other structural tissues, and sarcomas can arise in any of these types of tissues. As a result, there are many subtypes of sarcoma, which are classified based on the specific tissue and type of cell from which the tumor originates. Sarcomas are primary connective tissue tumors, meaning that they arise in connective tissues. This is in contrast to secondary connective tissue tumors, which occur when a cancer from elsewhere in the body spreads to the connective tissue. Sarcomas are one of five different types of cancer, classified by the cell type from which they originate. The word sarcoma is derived from the Greek σάρκωμα sarkōma 'fleshy excrescence or substance', itself from σάρξsarx meaning 'flesh'.

Imatinib, sold under the brand names Gleevec and Glivec (both marketed worldwide by Novartis) among others, is an oral targeted therapy medication used to treat cancer. Imatinib is a small molecule inhibitor targeting multiple tyrosine kinases such as CSF1R, ABL, c-KIT, FLT3, and PDGFR-β. Specifically, it is used for chronic myelogenous leukemia (CML) and acute lymphocytic leukemia (ALL) that are Philadelphia chromosome–positive (Ph+), certain types of gastrointestinal stromal tumors (GIST), hypereosinophilic syndrome (HES), chronic eosinophilic leukemia (CEL), systemic mastocytosis, and myelodysplastic syndrome.

Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract. GISTs arise in the smooth muscle pacemaker interstitial cell of Cajal, or similar cells. They are defined as tumors whose behavior is driven by mutations in the KIT gene (85%), PDGFRA gene (10%), or BRAF kinase (rare). 95% of GISTs stain positively for KIT (CD117). Most (66%) occur in the stomach and gastric GISTs have a lower malignant potential than tumors found elsewhere in the GI tract.

Fibrosarcoma is a malignant mesenchymal tumour derived from fibrous connective tissue and characterized by the presence of immature proliferating fibroblasts or undifferentiated anaplastic spindle cells in a storiform pattern. Fibrosarcomas mainly arise in people between the ages of 25 and 79. It originates in fibrous tissues of the bone and invades long or flat bones such as the femur, tibia, and mandible. It also involves the periosteum and overlying muscle.

Phyllodes tumors, are a rare type of biphasic fibroepithelial mass that form from the periductal stromal and epithelial cells of the breast. They account for less than 1% of all breast neoplasms. They were previously termed cystosarcoma phyllodes, coined by Johannes Müller in 1838, before being renamed to phyllodes tumor by the World Health Organization in 2003. Phullon, which means 'leaf' in Greek, describes the unique papillary projections characteristic of phyllodes tumors on histology. Diagnosis is made via a core-needle biopsy and treatment is typically surgical resection with wide margins (>1 cm), due to their propensity to recur.

Aggressive fibromatosis or desmoid tumor is a rare condition. Desmoid tumors are a type of fibromatosis and related to sarcoma, though without the ability to spread throughout the body (metastasize). The tumors arise from cells called fibroblasts, which are found throughout the body and provide structural support, protection to the vital organs, and play a critical role in wound healing. These tumors tend to occur in women in their thirties, but can occur in anyone at any age. They can be either relatively slow-growing or malignant. However, aggressive fibromatosis is locally aggressive and can cause life-threatening problems or even death when the tumors compress vital organs such as intestines, kidneys, lungs, blood vessels, or nerves. The condition is rarely fatal. Most cases are sporadic, but some are associated with familial adenomatous polyposis (FAP). Approximately 10% of individuals with Gardner's syndrome, a type of FAP with extracolonic features, have desmoid tumors.
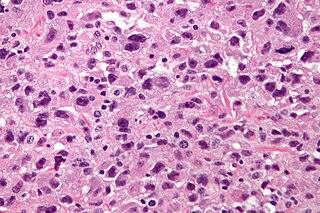
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO), 2020, as a rare, poorly differentiated neoplasm, i.e. an abnormal growth of cells that have an unclear identity and/or cell of origin. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers known or thought to derive from mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.
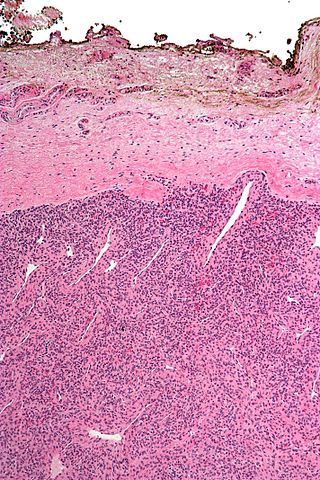
A resection margin or surgical margin is the margin of apparently non-tumorous tissue around a tumor that has been surgically removed, called "resected", in surgical oncology. The resection is an attempt to remove a cancer tumor so that no portion of the malignant growth extends past the edges or margin of the removed tumor and surrounding tissue. These are retained after the surgery and examined microscopically by a pathologist to see if the margin is indeed free from tumor cells. If cancerous cells are found at the edges the operation is much less likely to achieve the desired results.
Giant cell fibroblastoma (GCF) is a rare type of soft-tissue tumor marked by painless nodules in the dermis and subcutaneous tissue. These tumors may come back after surgery, but they do not spread to other parts of the body. They occur mostly in boys. GCF tumor tissues consist of bland spindle-shaped or stellate-shaped cells interspersed among multinucleated giant cells.
Collagenous fibroma, also known as desmoplastic fibroblastoma, is a slow-growing, deep-set, benign fibrous tumor, usually located in the deep subcutis, fascia, aponeurosis, or skeletal muscle of the extremities, limb girdles, or head and neck regions. The World Health Organization in 2020 reclassified desmoplastic fibroblastoma/collagenous fibroma as a specific benign tumor type within the broad category of fibroblastic and myofibroblastic tumors.

Koenen's tumor (KT), also commonly termed periungual angiofibroma, is a subtype of the angiofibromas. Angiofibromas are benign papule, nodule, and/or tumor lesions that are separated into various subtypes based primarily on the characteristic locations of their lesions. KTs are angiofibromas that develop in and under the toenails and/or fingernails. KTs were once considered as the same as another subtype of the angiofibromas viz., acral angiofibromas. While the literature may still sometimes regard KTs as acral angiofibromas, acral angiofibromas are characteristically located in areas close to but not in the toenails and fingernails as well as in the soles of the feet and palms of the hands. KTs are here regarded as distinct from acral angiofibromas.

Spiradenomas (SA) are rare, benign cutaneous adnexal tumors that may progress to become their malignant counterparts, i.e. spiradenocarcinomas (SAC). Cutaneous adnexal tumors are a group of skin tumors consisting of tissues that have differentiated towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands. SA and SAC tumors were regarded as eccrine gland tumors and termed eccrine spiradenomas and eccrine spiradenocarcinomas, respectively. However, more recent studies have found them to be hair follicle tumors and commonly term them spiradenomas and spiradenocarcinomas, respectively. Further confusing the situation, SA-like and SAC-like tumors are also 1) manifestations of the inherited disorder, CYLD cutaneous syndrome (CCS), and 2) have repeatedly been confused with an entirely different tumor, adenoid cystic carcinomas of the salivary gland. Here, SA and SAC are strictly defined as sporadic hair follicle tumors that do not include the hereditary CCS spiradenomas and heridtary spiradenocarcinoms of CCS or the adenoid cystic carcinomas.

Low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade sarcoma first described by H. L. Evans in 1987. LGFMS are soft tissue tumors of the mesenchyme-derived connective tissues; on microscopic examination, they are found to be composed of spindle-shaped cells that resemble fibroblasts. These fibroblastic, spindle-shaped cells are neoplastic cells that in most cases of LGFMS express fusion genes, i.e. genes composed of parts of two different genes that form as a result of mutations. The World Health Organization (2020) classified LGFMS as a specific type of tumor in the category of malignant fibroblastic and myofibroblastic tumors.
Andrea A. Hayes Dixon is an American surgeon. She was the first pediatric surgeon to perform a high-risk, life-saving procedure in children with a rare form of cancer and developed the first orthotropic xenograft model of metastatic Ewing's sarcoma. In 2002, she became the first African American female pediatric surgeon board-certified in the United States.
Fibroblastic and myofibroblastic tumors (FMTs) develop from the mesenchymal stem cells which differentiate into fibroblasts and/or the myocytes/myoblasts that differentiate into muscle cells. FMTs are a heterogeneous group of soft tissue neoplasms. The World Health Organization (2020) defined tumors as being FMTs based on their morphology and, more importantly, newly discovered abnormalities in the expression levels of key gene products made by these tumors' neoplastic cells. Histopathologically, FMTs consist of neoplastic connective tissue cells which have differented into cells that have microscopic appearances resembling fibroblasts and/or myofibroblasts. The fibroblastic cells are characterized as spindle-shaped cells with inconspicuous nucleoli that express vimentin, an intracellular protein typically found in mesenchymal cells, and CD34, a cell surface membrane glycoprotein. Myofibroblastic cells are plumper with more abundant cytoplasm and more prominent nucleoli; they express smooth muscle marker proteins such as smooth muscle actins, desmin, and caldesmon. The World Health Organization further classified FMTs into four tumor forms based on their varying levels of aggressiveness: benign, intermediate, intermediate, and malignant.
Lipofibromatosis-like neural tumor (LPF-NT) is an extremely rare soft tissue tumor first described by Agaram et al in 2016. As of mid-2021, at least 39 cases of LPF-NT have been reported in the literature. LPF-NT tumors have several features that resemble lipofibromatosis (LPF) tumors, malignant peripheral nerve sheath tumors, spindle cell sarcomas, low-grade neural tumors, peripheral nerve sheath tumors, and other less clearly defined tumors; Prior to the Agaram at al report, LPF-NTs were likely diagnosed as variants or atypical forms of these tumors. The analyses of Agaram at al and subsequent studies uncovered critical differences between LPF-NT and the other tumor forms which suggest that it is a distinct tumor entity differing not only from lipofibromatosis but also the other tumor forms.
Myxofibrosarcoma (MFS), although a rare type of tumor, is one of the most common soft tissue sarcomas, i.e. cancerous tumors, that develop in the soft tissues of elderly individuals. Initially considered to be a type of histiocytoma termed fibrous histiocytoma or myxoid variant of malignant fibrous histiocytoma, Angervall et al. termed this tumor myxofibrosarcoma in 1977. In 2020, the World Health Organization reclassified MFS as a separate and distinct tumor in the category of malignant fibroblastic and myofibroblastic tumors.
Sclerosing epithelioid fibrosarcoma (SEF) is a very rare malignant tumor of soft tissues that on microscopic examination consists of small round or ovoid neoplastic epithelioid fibroblast-like cells, i.e. cells that have features resembling both epithelioid cells and fibroblasts. In 2020, the World Health Organization classified SEF as a distinct tumor type in the category of malignant fibroblastic and myofibroblastic tumors. However, current studies have reported that low-grade fibromyxoid sarcoma (LGFMS) has many clinically and pathologically important features characteristic of SEF; these studies suggest that LGSFMS may be an early form of, and over time progress to become, a SEF. Since the World Health Organization has classified LGFMS as one of the malignant fibroblastic and myofibroblastic tumors that is distinctly different than SEF, SEF and LGFMS are here regarded as different tumor forms.
Dermatofibrosarcoma protuberans, fibrosarcomatous (DFSP-FS), also termed fibrosarcomatous dermatofibrosarcoma protuberans, is a rare type of tumor located in the dermis. DFSP-FS tumors have been viewed as: 1) a more aggressive form of the dermatofibrosarcoma protuberans (DFSP) tumors because they have areas that resemble and tend to behave like malignant fibrosarcomas or 2) as a distinctly different tumor than DFSP. DFSP-FS tumors are related to DFSP. For example, surgically removed DFSP tumors often recur with newly developed fibrobosarcoma-like areas. Nonetheless, the World Health Organization (WHO), 2020, classified DFSP and DFSP-FS as different tumors with DFSP being in the category of benign and DFSP-FS in the category of rarely metastasizing fibroblastic and myofibroblastic tumors. This article follows the WHO classification: the 5-15% of DFSP tumors that have any areas of fibrosarcomatous microscopic histopathology are here considered DFSP-FS rather than DFSP tumors.
Gardner fibroma (GF) is a benign fibroblastic tumor. GF tumors typically develop in the dermis and adjacent subcutaneous tissue lying just below the dermis. These tumors typically occur on the back, abdomen, and other superficial sites but in rare cases have been diagnoses in internal sites such as the retroperitoneum and around the large blood vessels in the upper thoracic cavity. The World Health Organization, 2020, classified Gardner fibroma as a benign tumor in the category of fibroblastic and myofibroblastic tumors.