
Robert G. Roeder is an American biochemist. He is known as a pioneer scientist in eukaryotic transcription. He discovered three distinct nuclear RNA polymerases in 1969 and characterized many proteins involved in the regulation of transcription, including basic transcription factors and the first mammalian gene-specific activator over five decades of research. He is the recipient of the Gairdner Foundation International Award in 2000, the Albert Lasker Award for Basic Medical Research in 2003, and the Kyoto Prize in 2021. He currently serves as Arnold and Mabel Beckman Professor and Head of the Laboratory of Biochemical and Molecular Biology at The Rockefeller University.
Glycoconjugates are the classification family for carbohydrates – referred to as glycans – which are covalently linked with chemical species such as proteins, peptides, lipids, and other compounds. Glycoconjugates are formed in processes termed glycosylation.

Catherine Dulac is a French–American biologist. She is the Higgins Professor in Molecular and Cellular Biology at Harvard University, where she served as department chair from 2007 to 2013. She is also an investigator at the Howard Hughes Medical Institute. She was born in 1963 in France. She came to the United States for her postdoctoral study in 1991.
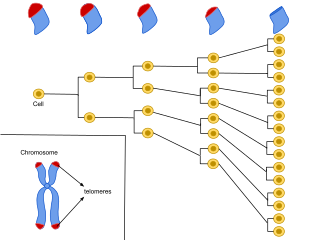
Cellular senescence is a phenomenon characterized by the cessation of cell division. In their experiments during the early 1960s, Leonard Hayflick and Paul Moorhead found that normal human fetal fibroblasts in culture reach a maximum of approximately 50 cell population doublings before becoming senescent. This process is known as "replicative senescence", or the Hayflick limit. Hayflick's discovery of mortal cells paved the path for the discovery and understanding of cellular aging molecular pathways. Cellular senescence can be initiated by a wide variety of stress inducing factors. These stress factors include both environmental and internal damaging events, abnormal cellular growth, oxidative stress, autophagy factors, among many other things.

RNA-Seq is a technique that uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA molecules in a biological sample, providing a snapshot of gene expression in the sample, also known as transcriptome.
Maternal to zygotic transition (MZT), also known as embryonic genome activation, is the stage in embryonic development during which development comes under the exclusive control of the zygotic genome rather than the maternal (egg) genome. The egg contains stored maternal genetic material mRNA which controls embryo development until the onset of MZT. After MZT the diploid embryo takes over genetic control. This requires both zygotic genome activation (ZGA), and degradation of maternal products. This process is important because it is the first time that the new embryonic genome is utilized and the paternal and maternal genomes are used in combination. The zygotic genome now drives embryo development.
MicroRNA sequencing (miRNA-seq), a type of RNA-Seq, is the use of next-generation sequencing or massively parallel high-throughput DNA sequencing to sequence microRNAs, also called miRNAs. miRNA-seq differs from other forms of RNA-seq in that input material is often enriched for small RNAs. miRNA-seq allows researchers to examine tissue-specific expression patterns, disease associations, and isoforms of miRNAs, and to discover previously uncharacterized miRNAs. Evidence that dysregulated miRNAs play a role in diseases such as cancer has positioned miRNA-seq to potentially become an important tool in the future for diagnostics and prognostics as costs continue to decrease. Like other miRNA profiling technologies, miRNA-Seq has both advantages and disadvantages.
Jeannie T. Lee is a Professor of Genetics at Harvard Medical School and the Massachusetts General Hospital, and a Howard Hughes Medical Institute Investigator. She is known for her work on X-chromosome inactivation and for discovering the functions of a new class of epigenetic regulators known as long noncoding RNAs (lncRNAs), including Xist and Tsix.

Bradley E. Bernstein is a biologist and Professor of Cell Biology at Harvard Medical School. He is Chair of the Department of Cancer Biology at the Dana–Farber Cancer Institute and the Director of the Broad Institute's Gene Regulation Observatory. He is known for contributions to the fields of epigenetics and cancer biology.

Nina Papavasiliou is an immunologist and Helmholtz Professor in the Division of Immune Diversity at the German Cancer Research Center in Heidelberg, Germany. She is also an adjunct professor at the Rockefeller University, where she was previously associate professor and head of the Laboratory of Lymphocyte Biology. She is best known for her work in the fields of DNA and RNA editing.

Trajectory inference or pseudotemporal ordering is a computational technique used in single-cell transcriptomics to determine the pattern of a dynamic process experienced by cells and then arrange cells based on their progression through the process. Single-cell protocols have much higher levels of noise than bulk RNA-seq, so a common step in a single-cell transcriptomics workflow is the clustering of cells into subgroups. Clustering can contend with this inherent variation by combining the signal from many cells, while allowing for the identification of cell types. However, some differences in gene expression between cells are the result of dynamic processes such as the cell cycle, cell differentiation, or response to an external stimuli. Trajectory inference seeks to characterize such differences by placing cells along a continuous path that represents the evolution of the process rather than dividing cells into discrete clusters. In some methods this is done by projecting cells onto an axis called pseudotime which represents the progression through the process.
CITE-Seq is a method for performing RNA sequencing along with gaining quantitative and qualitative information on surface proteins with available antibodies on a single cell level. So far, the method has been demonstrated to work with only a few proteins per cell. As such, it provides an additional layer of information for the same cell by combining both proteomics and transcriptomics data. For phenotyping, this method has been shown to be as accurate as flow cytometry by the groups that developed it. It is currently one of the main methods, along with REAP-Seq, to evaluate both gene expression and protein levels simultaneously in different species.
Richard William Carthew is a developmental biologist and quantitative biologist at Northwestern University. He is a professor of molecular biosciences and is the director of the NSF-Simons Center for Quantitative Biology.
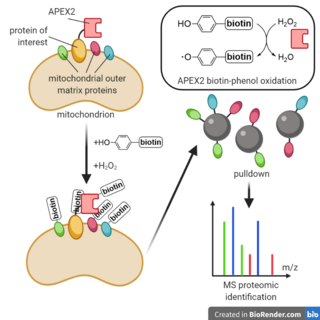
Enzyme-catalyzed proximity labeling (PL), also known as proximity-based labeling, is a laboratory technique that labels biomolecules, usually proteins or RNA, proximal to a protein of interest. By creating a gene fusion in a living cell between the protein of interest and an engineered labeling enzyme, biomolecules spatially proximal to the protein of interest can then be selectively marked with biotin for pulldown and analysis. Proximity labeling has been used for identifying the components of novel cellular structures and for determining protein-protein interaction partners, among other applications.
Bryan Richard Cullen is a James B. Duke Professor of Molecular Genetics and Microbiology at Duke University Medical Center in Durham, North Carolina. Cullen was the Founding Director of the Duke University Center for Virology.
Aging is characterized by a progressive loss of physiological integrity, leading to impaired function and increased vulnerability to death. The hallmarks of aging are the types of biochemical changes that occur in all organisms that experience biological aging and lead to a progressive loss of physiological integrity, impaired function and, eventually, death. They were first listed in a landmark paper in 2013 to conceptualize the essence of biological aging and its underlying mechanisms.
Deterministic Barcoding in Tissue for Spatial Omics Sequencing (DBiT-seq) was developed at Yale University by Rong Fan and colleagues in 2020 to create a multi-omics approach for studying spatial gene expression heterogenicity within a tissue sample. This method can be used for the co-mapping mRNA and protein levels at a near single-cell resolution in fresh or frozen formaldehyde-fixed tissue samples. DBiT-seq utilizes next generation sequencing (NGS) and microfluidics. This method allows for simultaneous spatial transcriptomic and proteomic analysis of a tissue sample. DBiT-seq improves upon previous spatial transcriptomics applications such as High-Definition Spatial Transcriptomics (HDST) and Slide-seq by increasing the number of detectable genes per pixel, increased cellular resolution, and ease of implementation.
Single-cell genome and epigenome by transposases sequencing (scGET-seq) is a DNA sequencing method for profiling open and closed chromatin. In contrast to single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq), which only targets active euchromatin. scGET-seq is also capable of probing inactive heterochromatin.
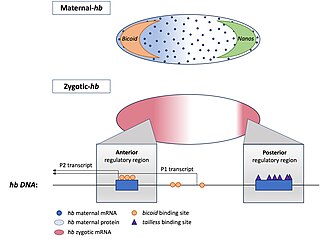
Hunchback is a maternal effect and zygotic gene expressed in the embryos of the fruit fly Drosophila melanogaster. In maternal effect genes, the RNA or protein from the mother’s gene is deposited into the oocyte or embryo before the embryo can express its own zygotic genes.
