Evolution and phylogenesis of Yersinia pestis
Yersinia pestis is a gram-negative bacterium and belongs to the family of Enterobatteriaceae. Its closest relatives are Yersinia pseudotuberculosis and Yersinia enterocolitica , which are environmental species.[ citation needed ]

They all possess the plasmid pCD1, which encodes for a type III secretory system. Among chromosomal protein-coding genes, their nucleotide genomic identity rates 97%. They are different in terms of their virulence potential and transmission mechanisms. [12]
Y. pestis is not a human-adapted bacteria. Its main reservoirs are rodents (like marmots, mice, great gerbils, voles and prairie dogs) and it is transmitted to humans by the flea. One of the most studied vectors of this pathogen is Xenopsylla cheopis .[ citation needed ]
After the bite of an infected flea, the bacteria enter into the host organism and travel to the closest lymph node, where bacteria replicate causing the large swellings called buboes. Bacteria can also disseminate into the bloodstream (causing septicaemia) and to the lung (causing pneumonia). The pulmonary disease has a direct human-to-human transmission. [13]
It has been determined that Y. pestis became so dangerous because of the acquisition of ymt (yersina murine toxin). This gene is present on the pMT1 plasmid and allowed the survival of the bacterium in the flea vector and facilitated colonization of the midgut in arthropod, giving rise to the past millennium large-scale pandemics. [13]
Early evolution and divergence from Yersinia pseudotuberculosis
Y. pestis is distinguishable from the other two species because of its pathogenicity and transmission mechanism. These differences are given by two plasmids: pPCP1, that confer to the bacterium its invasive properties in humans and pMT1, which is involved in flea colonisation (along with some loss of function on bacterial chromosomal genes).[ citation needed ]
Samples dated on the Late Neolithic and Bronze Age allowed identifying a first genetic divergence between Y. pseudotuberculosis and Y. pestis ancestors. The characteristics that confer to Y. pestis its virulence were absent in these strains: they lack of ymt, a gene necessary to the colonization of the vector; also, they presented an active form of genes required for biofilm formation (inactive in the pathogen Y. pestis) and an active flagellin gene, that is an inducer of immune response (is a pseudogene in Y. pestis). [1]
The comparison of a draft of the genome and the two plasmids (pCD1 and pMT1) with samples of Black Death victims (1348-1349) in the East Smithfield burial ground underlined a very high genetic conservation of the sequence: only 97 single-nucleotide differences over 660 years. [14]
Y. pestis microevolution
The London 6330 individual strain owns mutations absent in other isolates of the same period (1348-1350): the reason may be either the presence of multiple strains circulating in Europe at the same time or the microevolution of one single strain during the pandemic. [14]
Three major outbreaks of plague
There are three pandemic outbreaks of Y. Pestis:[ citation needed ]
- The first is known as the Plague of Justinian, it first occurred in Egypt in 541-543 and then spread to Constantinople and neighbouring regions. It had outbreaks in Europe until 750 CE. Phylogenetic analysis showed that both genomes belong to a lineage that is extinct today and is closely related to strains from modern-day China, which suggest the possibility of an East Asian origin of the first pandemic.
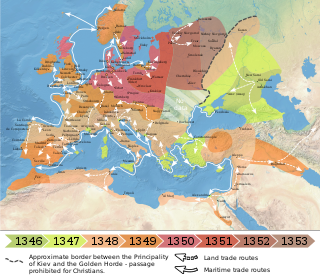
- The second pandemic is known as the Black Death or as Great Pestilence. It occurred in 1346-1352 in Europe and had a lot of resurgences of plague, it continued until the 18th century. It could be possible that in this pandemic there were two different strains of Y. pestis that entered the continent through different pulses.
- The third pandemic started in China in 1860. It has a fast spread to other countries, due to the use of railways and steamships.
The strains associated with the Justinian Plague appear on a novel branch, which is phylogenetically distinct from the second and the third plague pandemics. The first strain of Y. pestis found during the second outbreak survives and give rise to modern branch 1 strains associated with the third pandemic outbreak.[ citation needed ]
The first plague bacteria and the second and third plague strain have a common ancestor. [2]
Linkage between 2nd and 3rd pandemics
In a recent study, [15] genomes of Y. pestis from three samples resumed in Barcelona (deceased between 1300-1420), Ellwangen (between 1486-1627) and Bolgar city (between 1298-1388). The date of death of the individuals analysed was determined thanks to radiocarbon dates; the last one was confirmed by the presence of a coin produced only after the year 1362. Of 223 samples from 178 individuals, only one for each site had a suitable amount of DNA and was finally selected for the whole genome sequencing of the bacillus (through a genome-capture assay, using as a draft Y. pseudotuberculosis genome and pMT1 and pCD1 from Y. pestis).[ citation needed ]
The alignment with a Y. pestis phylogeny tree created with previously known ancient genomes revealed an increase genetic diversity outside of China in comparison to what was previously thought; all the three new genomes mapped in Branch 1 and possess two SNPs associated to the Black Death (all the genomes of Y. pestis dated to the Black Death map in Branch 1). [14] The Barcelona strain has no differences with the London strain; the two individuals from which the genome was obtained died of plague with a distance of some months (spring and autumn 1348 [16] [17] ) underlining the presence in Europe of a single wave of plague with low genetic diversity. The Ellwangen strain maps in a sub-branch of Branch 1 and is ancestral to a previously sequenced strain (L'Observance). [18] It descends from the one circulating in London and Barcelona during the Black Death but also have additional mutations. It is therefore considered a lineage diverged from Branch 1 before the 16th century (Ellwangen outbreak) and with no known modern descendants. [15]
In comparison with isolates from the Black Death, the Bolgar city strain presents:
- p3 and p4, shared by the "London individual 6330";
- p6, shared with all modern Branch 1 strains;
- p7, unique of this strain;
The Bolgar City strain possesses SNPs associated to the Black Death and can be an evidence of a movement of plague towards east; These findings give credit to one of the models that try to explain the linkage between 2nd and 3rd pandemic: in this scenario, there was a single exit of plague to Europe (causing the Black Death) that after a radiation event, travelled eastward to establish in former soviet union and Asia, from which it spread in the 18th century to give raise to the 3rd pandemic. [15]
Another hypothesis is that the 3rd pandemic's lineage may have been generated by a pre-existing genetic variability in Y. pestis strains in China: this hypothesis is actually supported by the correlation between following waves of the pandemic in Europe and climatic fluctuations that would have allowed its spread in the continent. This model can't explain the genetic diversity of the Black Death (four different lineages at least, that would have required the introduction from Asia of four different strains). [19]
Again, there are two models that try to explain the multiple plague outbreaks in Europe following the black death:
- Repeated introduction of plague from Asia. This scenario is compatible with the 2nd theory discussed before that sees a genetic variability of Y. pestis in China; [20]
- Presence in Europe of a reservoir (now extinct) that caused continue outbreaks until the 18th century;
Both models can be valid and nowadays we're not able to demonstrate one over the other. However, the Ellwangen strain genome sequenced in this study may be considered a proof of the second hypothesis due to the geographical position of the city that tends to exclude the possibility of an introduction of plague from eastward. [15]
Modern Y. pestis strains
Sequencing of Y. pestis genomes allowed to discover a variation event preceding Black Death that gave rise to many strains that circulate today. [15]