In genetics, shotgun sequencing is a method used for sequencing random DNA strands. It is named by analogy with the rapidly expanding, quasi-random shot grouping of a shotgun.
A bacterial artificial chromosome (BAC) is a DNA construct, based on a functional fertility plasmid, used for transforming and cloning in bacteria, usually E. coli. F-plasmids play a crucial role because they contain partition genes that promote the even distribution of plasmids after bacterial cell division. The bacterial artificial chromosome's usual insert size is 150–350 kbp. A similar cloning vector called a PAC has also been produced from the DNA of P1 bacteriophage.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology cannot read whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically the short fragments, called reads, result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
This is a list of topics in molecular biology. See also index of biochemistry articles.
Primer walking, also called chromosome walking, is a technique used to clone a gene from its known closest markers. As a result, it is employed in cloning and sequencing efforts in plants, fungi, and mammals with minor alterations. This technique, also known as "directed sequencing," employs a series of Sanger sequencing reactions to either confirm the reference sequence of a known plasmid or PCR product based on the reference sequence or to discover the unknown sequence of a full plasmid or PCR product by designing primers to sequence overlapping sections.

Sanger sequencing is a method of DNA sequencing that involves electrophoresis and is based on the random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. After first being developed by Frederick Sanger and colleagues in 1977, it became the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by next generation sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use for smaller-scale projects and for validation of deep sequencing results. It still has the advantage over short-read sequencing technologies in that it can produce DNA sequence reads of >500 nucleotides and maintains a very low error rate with accuracies around 99.99%. Sanger sequencing is still actively being used in efforts for public health initiatives such as sequencing the spike protein from SARS-CoV-2 as well as for the surveillance of norovirus outbreaks through the Center for Disease Control and Prevention's (CDC) CaliciNet surveillance network.
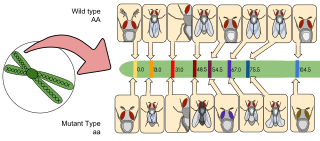
Gene mapping describes the methods used to identify the locus of a gene and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.
A genomic library is a collection of the total genomic DNA from a single organism. The DNA is stored in a population of identical vectors, each containing a different insert of DNA. In order to construct a genomic library, the organism's DNA is extracted from cells and then digested with a restriction enzyme to cut the DNA into fragments of a specific size. The fragments are then inserted into the vector using DNA ligase. Next, the vector DNA can be taken up by a host organism - commonly a population of Escherichia coli or yeast - with each cell containing only one vector molecule. Using a host cell to carry the vector allows for easy amplification and retrieval of specific clones from the library for analysis.

Artificial gene synthesis, or simply gene synthesis, refers to a group of methods that are used in synthetic biology to construct and assemble genes from nucleotides de novo. Unlike DNA synthesis in living cells, artificial gene synthesis does not require template DNA, allowing virtually any DNA sequence to be synthesized in the laboratory. It comprises two main steps, the first of which is solid-phase DNA synthesis, sometimes known as DNA printing. This produces oligonucleotide fragments that are generally under 200 base pairs. The second step then involves connecting these oligonucleotide fragments using various DNA assembly methods. Because artificial gene synthesis does not require template DNA, it is theoretically possible to make a completely synthetic DNA molecule with no limits on the nucleotide sequence or size.
DNA sequencing theory is the broad body of work that attempts to lay analytical foundations for determining the order of specific nucleotides in a sequence of DNA, otherwise known as DNA sequencing. The practical aspects revolve around designing and optimizing sequencing projects, predicting project performance, troubleshooting experimental results, characterizing factors such as sequence bias and the effects of software processing algorithms, and comparing various sequencing methods to one another. In this sense, it could be considered a branch of systems engineering or operations research. The permanent archive of work is primarily mathematical, although numerical calculations are often conducted for particular problems too. DNA sequencing theory addresses physical processes related to sequencing DNA and should not be confused with theories of analyzing resultant DNA sequences, e.g. sequence alignment. Publications sometimes do not make a careful distinction, but the latter are primarily concerned with algorithmic issues. Sequencing theory is based on elements of mathematics, biology, and systems engineering, so it is highly interdisciplinary. The subject may be studied within the context of computational biology.
Optical mapping is a technique for constructing ordered, genome-wide, high-resolution restriction maps from single, stained molecules of DNA, called "optical maps". By mapping the location of restriction enzyme sites along the unknown DNA of an organism, the spectrum of resulting DNA fragments collectively serves as a unique "fingerprint" or "barcode" for that sequence. Originally developed by Dr. David C. Schwartz and his lab at NYU in the 1990s this method has since been integral to the assembly process of many large-scale sequencing projects for both microbial and eukaryotic genomes. Later technologies use DNA melting, DNA competitive binding or enzymatic labelling in order to create the optical mappings.
Polony sequencing is an inexpensive but highly accurate multiplex sequencing technique that can be used to “read” millions of immobilized DNA sequences in parallel. This technique was first developed by Dr. George Church's group at Harvard Medical School. Unlike other sequencing techniques, Polony sequencing technology is an open platform with freely downloadable, open source software and protocols. Also, the hardware of this technique can be easily set up with a commonly available epifluorescence microscopy and a computer-controlled flowcell/fluidics system. Polony sequencing is generally performed on paired-end tags library that each molecule of DNA template is of 135 bp in length with two 17–18 bp paired genomic tags separated and flanked by common sequences. The current read length of this technique is 26 bases per amplicon and 13 bases per tag, leaving a gap of 4–5 bases in each tag.

In bioinformatics, hybrid genome assembly refers to utilizing various sequencing technologies to achieve the task of assembling a genome from fragmented, sequenced DNA resulting from shotgun sequencing. Genome assembly presents one of the most challenging tasks in genome sequencing as most modern DNA sequencing technologies can only produce reads that are, on average, 25-300 base pairs in length. This is orders of magnitude smaller than the average size of a genome. This assembly is computationally difficult and has some inherent challenges, one of these challenges being that genomes often contain complex tandem repeats of sequences that can be thousands of base pairs in length. These repeats can be long enough that second generation sequencing reads are not long enough to bridge the repeat, and, as such, determining the location of each repeat in the genome can be difficult. Resolving these tandem repeats can be accomplished by utilizing long third generation sequencing reads, such as those obtained using the PacBio RS DNA sequencer. These sequences are, on average, 10,000-15,000 base pairs in length and are long enough to span most repeated regions. Using a hybrid approach to this process can increase the fidelity of assembling tandem repeats by being able to accurately place them along a linear scaffold and make the process more computationally efficient.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.

Jumping libraries or junction-fragment libraries are collections of genomic DNA fragments generated by chromosome jumping. These libraries allow the analysis of large areas of the genome and overcome distance limitations in common cloning techniques. A jumping library clone is composed of two stretches of DNA that are usually located many kilobases away from each other. The stretch of DNA located between these two "ends" is deleted by a series of biochemical manipulations carried out at the start of this cloning technique.
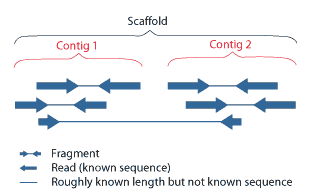
Scaffolding is a technique used in bioinformatics. It is defined as follows:
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.

End-sequence profiling (ESP) is a method based on sequence-tagged connectors developed to facilitate de novo genome sequencing to identify high-resolution copy number and structural aberrations such as inversions and translocations.
Synthetic genome is a synthetically built genome whose formation involves either genetic modification on pre-existing life forms or artificial gene synthesis to create new DNA or entire lifeforms. The field that studies synthetic genomes is called synthetic genomics.
A plant genome assembly represents the complete genomic sequence of a plant species, which is assembled into chromosomes and other organelles by using DNA fragments that are obtained from different types of sequencing technology.
Physical map is a technique used in molecular biology to find the order and physical distance between DNA base pairs by DNA markers. It is one of the gene mapping techniques which can determine the sequence of DNA base pairs with high accuracy. Genetic mapping, another approach of gene mapping, can provide markers needed for the physical mapping. However, as the former deduces the relative gene position by recombination frequencies, it is less accurate than the latter.
