
Chromosomal crossover, or crossing over, is the exchange of genetic material during sexual reproduction between two homologous chromosomes' non-sister chromatids that results in recombinant chromosomes. It is one of the final phases of genetic recombination, which occurs in the pachytene stage of prophase I of meiosis during a process called synapsis. Synapsis begins before the synaptonemal complex develops and is not completed until near the end of prophase I. Crossover usually occurs when matching regions on matching chromosomes break and then reconnect to the other chromosome.

DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encodes its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in tens of thousands of individual molecular lesions per cell per day. Many of these lesions cause structural damage to the DNA molecule and can alter or eliminate the cell's ability to transcribe the gene that the affected DNA encodes. Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. As a consequence, the DNA repair process is constantly active as it responds to damage in the DNA structure. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur. This can eventually lead to malignant tumors, or cancer as per the two-hit hypothesis.
RecQ helicase is a family of helicase enzymes initially found in Escherichia coli that has been shown to be important in genome maintenance. They function through catalyzing the reaction ATP + H2O → ADP + P and thus driving the unwinding of paired DNA and translocating in the 3' to 5' direction. These enzymes can also drive the reaction NTP + H2O → NDP + P to drive the unwinding of either DNA or RNA.

Non-homologous end joining (NHEJ) is a pathway that repairs double-strand breaks in DNA. It is called "non-homologous" because the break ends are directly ligated without the need for a homologous template, in contrast to homology directed repair (HDR), which requires a homologous sequence to guide repair. NHEJ is active in both non-dividing and proliferating cells, while HDR is not readily accessible in non-dividing cells. The term "non-homologous end joining" was coined in 1996 by Moore and Haber.

A heteroduplex is a double-stranded (duplex) molecule of nucleic acid originated through the genetic recombination of single complementary strands derived from different sources, such as from different homologous chromosomes or even from different organisms.
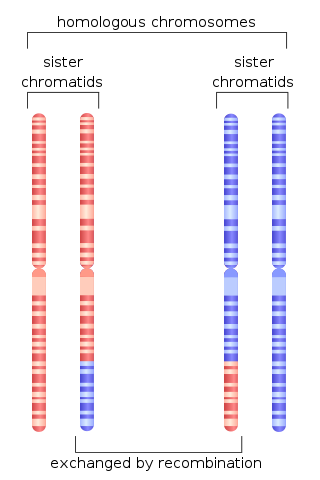
Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids.
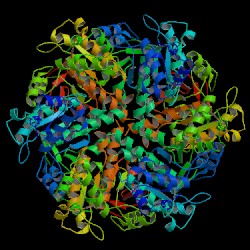
DNA repair protein RAD51 homolog 1 is a protein encoded by the gene RAD51. The enzyme encoded by this gene is a member of the RAD51 protein family which assists in repair of DNA double strand breaks. RAD51 family members are homologous to the bacterial RecA, Archaeal RadA and yeast Rad51. The protein is highly conserved in most eukaryotes, from yeast to humans.

Nibrin, also known as NBN or NBS1, is a protein which in humans is encoded by the NBN gene.

Poly [ADP-ribose] polymerase 1 (PARP-1) also known as NAD+ ADP-ribosyltransferase 1 or poly[ADP-ribose] synthase 1 is an enzyme that in humans is encoded by the PARP1 gene. It is the most abundant of the PARP family of enzymes, accounting for 90% of the NAD+ used by the family. PARP1 is mostly present in cell nucleus, but cytosolic fraction of this protein was also reported.

Tumor suppressor p53-binding protein 1 also known as p53-binding protein 1 or 53BP1 is a protein that in humans is encoded by the TP53BP1 gene.

RAD52 homolog , also known as RAD52, is a protein which in humans is encoded by the RAD52 gene.

DNA repair and recombination protein RAD54-like is a protein that in humans is encoded by the RAD54L gene.

E3 ubiquitin-protein ligase FANCL is an enzyme that in humans is encoded by the FANCL gene.
BRCA1-A complex subunit BRE is a protein that in humans is encoded by the BRE gene.
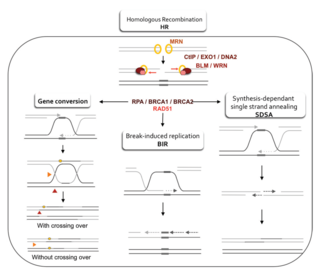
Homology-directed repair (HDR) is a mechanism in cells to repair double-strand DNA lesions. The most common form of HDR is homologous recombination. The HDR mechanism can only be used by the cell when there is a homologous piece of DNA present in the nucleus, mostly in G2 and S phase of the cell cycle. Other examples of homology-directed repair include single-strand annealing and breakage-induced replication. When the homologous DNA is absent, another process called non-homologous end joining (NHEJ) takes place instead.
Microhomology-mediated end joining (MMEJ), also known as alternative nonhomologous end-joining (Alt-NHEJ) is one of the pathways for repairing double-strand breaks in DNA. As reviewed by McVey and Lee, the foremost distinguishing property of MMEJ is the use of microhomologous sequences during the alignment of broken ends before joining, thereby resulting in deletions flanking the original break. MMEJ is frequently associated with chromosome abnormalities such as deletions, translocations, inversions and other complex rearrangements.
Synthesis-dependent strand annealing (SDSA) is a major mechanism of homology-directed repair of DNA double-strand breaks (DSBs). Although many of the features of SDSA were first suggested in 1976, the double-Holliday junction model proposed in 1983 was favored by many researchers. In 1994, studies of double-strand gap repair in Drosophila were found to be incompatible with the double-Holliday junction model, leading researchers to propose a model they called synthesis-dependent strand annealing. Subsequent studies of meiotic recombination in S. cerevisiae found that non-crossover products appear earlier than double-Holliday junctions or crossover products, challenging the previous notion that both crossover and non-crossover products are produced by double-Holliday junctions and leading the authors to propose that non-crossover products are generated through SDSA.
Lumír Krejčí is a Czech biochemist. His research is focused on regulatory processes involved in maintaining genome integrity. Currently, as Associate Professor in biochemistry, he leads the laboratory of recombination and DNA repair (LORD) at the Department of Biology, Faculty of Medicine, at Masaryk University in Brno.
Telomeres, the caps on the ends of eukaryotic chromosomes, play critical roles in cellular aging and cancer. An important facet to how telomeres function in these roles is their involvement in cell cycle regulation.

DNA end resection, also called 5′–3′ degradation, is a biochemical process where the blunt end of a section of double-stranded DNA (dsDNA) is modified by cutting away some nucleotides from the 5' end to produce a 3' single-stranded sequence. The presence of a section of single-stranded DNA (ssDNA) allows the broken end of the DNA to line up accurately with a matching sequence, so that it can be accurately repaired.


