Agarose gel electrophoresis is a method of gel electrophoresis used in biochemistry, molecular biology, genetics, and clinical chemistry to separate a mixed population of macromolecules such as DNA or proteins in a matrix of agarose, one of the two main components of agar. The proteins may be separated by charge and/or size, and the DNA and RNA fragments by length. Biomolecules are separated by applying an electric field to move the charged molecules through an agarose matrix, and the biomolecules are separated by size in the agarose gel matrix.

Gel electrophoresis is a method for separation and analysis of biomacromolecules and their fragments, based on their size and charge. It is used in clinical chemistry to separate proteins by charge or size and in biochemistry and molecular biology to separate a mixed population of DNA and RNA fragments by length, to estimate the size of DNA and RNA fragments or to separate proteins by charge.
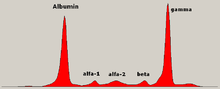
The isoelectric point (pI, pH(I), IEP), is the pH at which a molecule carries no net electrical charge or is electrically neutral in the statistical mean. The standard nomenclature to represent the isoelectric point is pH(I). However, pI is also used. For brevity, this article uses pI. The net charge on the molecule is affected by pH of its surrounding environment and can become more positively or negatively charged due to the gain or loss, respectively, of protons (H+).
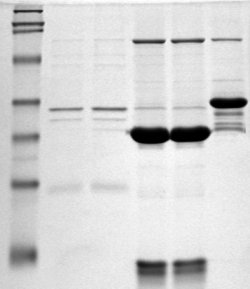
Polyacrylamide gel electrophoresis (PAGE) is a technique widely used in biochemistry, forensic chemistry, genetics, molecular biology and biotechnology to separate biological macromolecules, usually proteins or nucleic acids, according to their electrophoretic mobility. Electrophoretic mobility is a function of the length, conformation, and charge of the molecule. Polyacrylamide gel electrophoresis is a powerful tool used to analyze RNA samples. When polyacrylamide gel is denatured after electrophoresis, it provides information on the sample composition of the RNA species.

The western blot, or western blotting, is a widely used analytical technique in molecular biology and immunogenetics to detect specific proteins in a sample of tissue homogenate or extract. Besides detecting the proteins, this technique is also utilized to visualize, distinguish, and quantify the different proteins in a complicated protein combination.

Gel electrophoresis of nucleic acids is an analytical technique to separate DNA or RNA fragments by size and reactivity. Nucleic acid molecules are placed on a gel, where an electric field induces the nucleic acids to migrate toward the positively charged anode. The molecules separate as they travel through the gel based on the each molecule's size and shape. Longer molecules move more slowly because they the gel resists their movement more forcefully than it resists shorter molecules. After some time, the electricity is turned off and the positions of the different molecules are analyzed.

Two-dimensional gel electrophoresis, abbreviated as 2-DE or 2-D electrophoresis, is a form of gel electrophoresis commonly used to analyze proteins. Mixtures of proteins are separated by two properties in two dimensions on 2D gels. 2-DE was first independently introduced by O'Farrell and Klose in 1975.
Protein purification is a series of processes intended to isolate one or a few proteins from a complex mixture, usually cells, tissues or whole organisms. Protein purification is vital for the specification of the function, structure and interactions of the protein of interest. The purification process may separate the protein and non-protein parts of the mixture, and finally separate the desired protein from all other proteins. Ideally, to study a protein of interest, it must be separated from other components of the cell so that contaminants will not interfere in the examination of the protein of interest's structure and function. Separation of one protein from all others is typically the most laborious aspect of protein purification. Separation steps usually exploit differences in protein size, physico-chemical properties, binding affinity and biological activity. The pure result may be termed protein isolate.
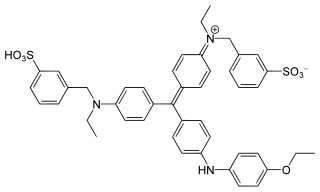
Coomassie brilliant blue is the name of two similar triphenylmethane dyes that were developed for use in the textile industry but are now commonly used for staining proteins in analytical biochemistry. Coomassie brilliant blue G-250 differs from Coomassie brilliant blue R-250 by the addition of two methyl groups. The name "Coomassie" is a registered trademark of Imperial Chemical Industries.

Isoelectric focusing (IEF), also known as electrofocusing, is a technique for separating different molecules by differences in their isoelectric point (pI). It is a type of zone electrophoresis usually performed on proteins in a gel that takes advantage of the fact that overall charge on the molecule of interest is a function of the pH of its surroundings.
Capillary electrophoresis (CE) is a family of electrokinetic separation methods performed in submillimeter diameter capillaries and in micro- and nanofluidic channels. Very often, CE refers to capillary zone electrophoresis (CZE), but other electrophoretic techniques including capillary gel electrophoresis (CGE), capillary isoelectric focusing (CIEF), capillary isotachophoresis and micellar electrokinetic chromatography (MEKC) belong also to this class of methods. In CE methods, analytes migrate through electrolyte solutions under the influence of an electric field. Analytes can be separated according to ionic mobility and/or partitioning into an alternate phase via non-covalent interactions. Additionally, analytes may be concentrated or "focused" by means of gradients in conductivity and pH.
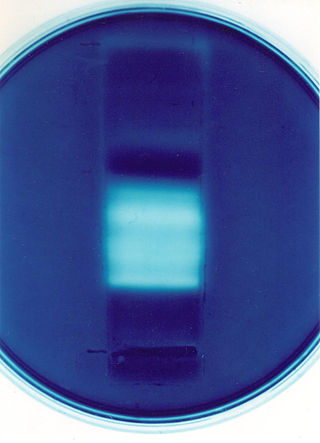
Zymography is an electrophoretic technique for the detection of hydrolytic enzymes, based on the substrate repertoire of the enzyme. Three types of zymography are used; in gel zymography, in situ zymography and in vivo zymography. For instance, gelatin embedded in a polyacrylamide gel will be digested by active gelatinases run through the gel. After Coomassie staining, areas of degradation are visible as clear bands against a darkly stained background.
In pathology, silver staining is the use of silver to selectively alter the appearance of a target in microscopy of histological sections; in temperature gradient gel electrophoresis; and in polyacrylamide gels.
A molecular-weight size marker, also referred to as a protein ladder, DNA ladder, or RNA ladder, is a set of standards that are used to identify the approximate size of a molecule run on a gel during electrophoresis, using the principle that molecular weight is inversely proportional to migration rate through a gel matrix. Therefore, when used in gel electrophoresis, markers effectively provide a logarithmic scale by which to estimate the size of the other fragments.
QPNC-PAGE, or QuantitativePreparativeNativeContinuousPolyacrylamideGel Electrophoresis, is a bioanalytical, one-dimensional, high-resolution and high-precision electrophoresis technique applied in biochemistry and bioinorganic chemistry to separate proteins quantitatively by isoelectric point and by continuous elution from a gel column.

An electrophoretic color marker is a chemical used to monitor the progress of agarose gel electrophoresis and polyacrylamide gel electrophoresis (PAGE) since DNA, RNA, and most proteins are colourless. The color markers are made up of a mixture of dyes that migrate through the gel matrix alongside the sample of interest. They are typically designed to have different mobilities from the sample components and to generate colored bands that can be used to assess the migration and separation of sample components.

Affinity electrophoresis is a general name for many analytical methods used in biochemistry and biotechnology. Both qualitative and quantitative information may be obtained through affinity electrophoresis. Cross electrophoresis, the first affinity electrophoresis method, was created by Nakamura et al. Enzyme-substrate complexes have been detected using cross electrophoresis. The methods include the so-called electrophoretic mobility shift assay, charge shift electrophoresis and affinity capillary electrophoresis. The methods are based on changes in the electrophoretic pattern of molecules through biospecific interaction or complex formation. The interaction or binding of a molecule, charged or uncharged, will normally change the electrophoretic properties of a molecule. Membrane proteins may be identified by a shift in mobility induced by a charged detergent. Nucleic acids or nucleic acid fragments may be characterized by their affinity to other molecules. The methods have been used for estimation of binding constants, as for instance in lectin affinity electrophoresis or characterization of molecules with specific features like glycan content or ligand binding. For enzymes and other ligand-binding proteins, one-dimensional electrophoresis similar to counter electrophoresis or to "rocket immunoelectrophoresis", affinity electrophoresis may be used as an alternative quantification of the protein. Some of the methods are similar to affinity chromatography by use of immobilized ligands.
Free-flow electrophoresis (FFE), also known as carrier-free electrophoresis, is a matrix-free, high-voltage electrophoretic separation technique. FFE is an analogous technique to capillary electrophoresis, with a comparable resolution, that can be used for scientific questions, where semi-preparative and preparative amounts of samples are needed. It is used to quantitatively separate samples according to differences in charge or isoelectric point by forming a pH gradient. Because of the versatility of the technique, a wide range of protocols for the separation of samples like rare metal ions, protein isoforms, multiprotein complexes, peptides, organelles, cells, DNA origami, blood serum and nanoparticles exist. The advantage of FFE is the fast and gentle separation of samples dissolved in a liquid solvent without any need of a matrix, like polyacrylamide in gel electrophoresis. This ensures a very high recovery rate since analytes do not adhere to any carrier or matrix structure. Because of its continuous nature and high volume throughput, this technique allows a fast separation of preparative amounts of samples with a very high resolution. Furthermore, the separations can be conducted under native or denaturing conditions.

Discontinuous electrophoresis is a type of polyacrylamide gel electrophoresis. It was developed by Ornstein and Davis. This method produces high resolution and good band definition. It is widely used technique for separating proteins according to size and charge.
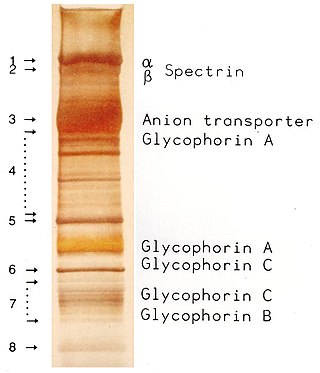
SDS-PAGE is a discontinuous electrophoretic system developed by Ulrich K. Laemmli which is commonly used as a method to separate proteins with molecular masses between 5 and 250 kDa. The combined use of sodium dodecyl sulfate and polyacrylamide gel eliminates the influence of structure and charge, and proteins are separated by differences in their size. At least up to 2012, the publication describing it was the most frequently cited paper by a single author, and the second most cited overall.