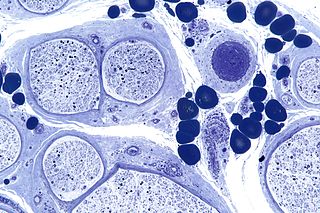
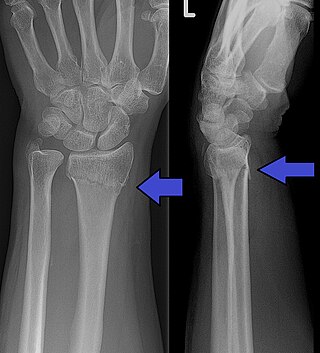
Hereditary sensory and autonomic neuropathy (HSAN) or hereditary sensory neuropathy (HSN) is a condition used to describe any of the types of this disease[1] which inhibit sensation.
Eight different clinical entities have been described under hereditary sensory and autonomic neuropathies – all characterized by progressive loss of function that predominantly affects the peripheral sensory nerves. Their incidence has been estimated to be about 1 in 250,000.
Hereditary sensory neuropathy type 1 is a condition characterized by nerve abnormalities in the legs and feet (peripheral neuropathy). Many people with this condition have tingling, weakness, and a reduced ability to feel pain and sense hot and cold. Some affected individuals do not lose sensation, but instead feel shooting pains in their legs and feet. As the disorder progresses, the sensory abnormalities can affect the hands, arms, shoulders, and abdomen. Affected individuals may also experience muscle wasting and weakness as they get older, but this varies widely within families.
Affected individuals typically get open sores (ulcers) on their feet or hands or infections of the soft tissue of the fingertips (whitlows) that are slow to heal. Because affected individuals cannot feel the pain of these sores, they may not seek treatment right away. Without treatment, the ulcers can become infected and may require amputation of the surrounding area.
Albeit rarely, people with hereditary sensory neuropathy type 1 may develop hearing loss caused by abnormalities of the inner ear (sensorineural hearing loss).
The signs and symptoms of hereditary sensory neuropathy type 1 typically appear during a person's teens or twenties. While the features of this disorder tend to worsen over time, affected individuals have a normal life expectancy if signs and symptoms are properly treated.
Type 1 is the most common form among the 5 types of HSAN. Its historical names include mal perforant du pied, ulcero-mutilating neuropathy, hereditary perforating ulcers, familial trophoneurosis, familial syringomyelia, hereditary sensory radicular neuropathy, among others.[3] This type includes a popular disease Charcot-Marie-Tooth type 2B syndrome (HMSN 2B).[3] that is also named as HSAN sub-type 1C.
Type 1 is inherited as an autosomal dominant trait. The disease usually starts during early adolescence or adulthood. The disease is characterized by the loss of pain sensation mainly in the distal parts of the lower limbs; that is, in the parts of the legs farther away from the center of the body. Since the affected individuals cannot feel pain, minor injuries in this area may not be immediately recognized and may develop into extensive ulcerations. Once infection occurs, further complications such as progressive destruction of underlying bones may follow and may necessitate amputation. In rare cases, the disease is accompanied with nerve deafness and muscle wasting. Autonomic disturbance, if present, appears as anhidrosis, a sweating abnormality. Examinations of the nerve structure and function showed signs of neuronal degeneration such as a marked reduction in the number of myelinated fibers and axonal loss. Sensory neurons lose the ability to transmit signals, while motor neurons has reduced ability to transmit signals.[3]
Genes related to Hereditary sensory and autonomic neuropathy Type 1:
Mutations in the SPTLC1 gene cause hereditary sensory neuropathy type 1. The SPTLC1 gene provides instructions for making one part (subunit) of an enzyme called serine palmitoyltransferase (SPT). The SPT enzyme is involved in making certain fats called sphingolipids. Sphingolipids are important components of cell membranes and play a role in many cell functions.
SPTLC1 gene mutations reduce the amount of SPTLC1 subunit that is produced and result in an SPT enzyme with decreased function. A lack of functional SPT enzyme leads to a decrease in sphingolipid production and a harmful buildup of certain byproducts. Sphingolipids are found in myelin, which is the covering that protects nerves and promotes the efficient transmission of nerve impulses. A decrease in sphingolipids disrupts the formation of myelin, causing nerve cells to become less efficient and eventually die. When sphingolipids are not made, an accumulation of toxic byproducts can also lead to nerve cell death. This gradual destruction of nerve cells results in loss of sensation and muscle weakness in people with hereditary sensory neuropathy type 1.
Type 2, Congenital sensory neuropathy
Hereditary sensory and autonomic neuropathy type II (HSAN2) is a condition that primarily affects the sensory nerve cells (sensory neurons) which transmit information about sensations such as pain, temperature, and touch. These sensations are impaired in people with HSAN2. In some affected people, the condition may also cause mild abnormalities of the autonomic nervous system, which controls involuntary body functions such as heart rate, digestion, and breathing. The signs and symptoms of HSAN2 typically begin in infancy or early childhood.
The first sign of HSAN2 is usually numbness in the hands and feet. Soon after, affected individuals lose the ability to feel pain or sense hot and cold. People with HSAN2 often develop open sores (ulcers) on their hands and feet. Because affected individuals cannot feel the pain of these sores, they may not seek treatment right away. Without treatment, the ulcers can become infected and may lead to amputation of the affected area. Unintentional self-injury is common in people with HSAN2, typically by biting the tongue, lips, or fingers. These injuries may lead to spontaneous amputation of the affected areas. Affected individuals often have injuries and fractures in their hands, feet, limbs, and joints that go untreated because of the inability to feel pain. Repeated injury can lead to a condition called Charcot joints, in which the bones and tissue surrounding joints are destroyed.
The effects of HSAN2 on the autonomic nervous system are more variable. Some infants with HSAN2 have trouble sucking, which makes it difficult for them to eat. People with HSAN2 may experience episodes in which breathing slows or stops for short periods (apnea); digestive problems such as the backflow of stomach acids into the esophagus (gastroesophageal reflux); or slow eye blink or gag reflexes. Affected individuals may also have weak deep tendon reflexes, such as the reflex being tested when a doctor taps the knee with a hammer.
Some people with HSAN2 experience a diminished sense of taste due to the loss of a type of taste bud on the tip of the tongue called lingual fungiform papillae.
Type 2, congenital sensory neuropathy (also historically known as Morvan's disease[4]), is characterized by onset of symptoms in early infancy or childhood. Upper & lower extremities are affected with chronic ulcerations and multiple injuries to fingers and feet. Pain sensation is affected predominantly and deep tendon reflexes are reduced. Autoamputation of the distal phalanges is common and so is neuropathic joint degeneration. The NCV shows reduced or absent sensory nerve action potentials and nerve biopsy shows total loss of myelinated fibers and reduced numbers of unmyelinated fibers. It is inherited as an autosomal recessive condition.
Genes related to Hereditary sensory and autonomic neuropathy Type 2:
There are two types of HSAN2, called HSAN2A and HSAN2B, each caused by mutations in a different gene. HSAN2A is caused by mutations in the WNK1 gene, and HSAN2B is caused by mutations in the RETREG1 gene (FAM134B). Although two different genes are involved, the signs and symptoms of HSAN2A and HSAN2B are the same.
The WNK1 gene provides instructions for making multiple versions (isoforms) of the WNK1 protein. HSAN2A is caused by mutations that affect a particular isoform called the WNK1/HSN2 protein. This protein is found in the cells of the nervous system, including nerve cells that transmit the sensations of pain, temperature, and touch (sensory neurons). The mutations involved in HSAN2A result in an abnormally short WNK1/HSN2 protein. Although the function of this protein is unknown, it is likely that the abnormally short version cannot function properly. People with HSAN2A have a reduction in the number of sensory neurons; however, the role that WNK1/HSN2 mutations play in that loss is unclear.
HSAN2B is caused by mutations in the RETREG1 gene. These mutations may lead to an abnormally short and nonfunctional protein. The RETREG1 protein is found in sensory and autonomic neurons. It is involved in the survival of neurons, particularly those that transmit pain signals, which are called nociceptive neurons. When the RETREG1 protein is nonfunctional, neurons die by a process of self-destruction called apoptosis.
The loss of neurons leads to the inability to feel pain, temperature, and touch sensations and to the impairment of the autonomic nervous system seen in people with HSAN2.
Familial dysautonomia is a genetic disorder that affects the development and survival of certain nerve cells. The disorder disturbs cells in the autonomic nervous system, which controls involuntary actions such as digestion, breathing, production of tears, and the regulation of blood pressure and body temperature. It also affects the sensory nervous system, which controls activities related to the senses, such as taste and the perception of pain, heat, and cold. Familial dysautonomia is also called hereditary sensory and autonomic neuropathy, type III.
Problems related to this disorder first appear during infancy. Early signs and symptoms include poor muscle tone (hypotonia), feeding difficulties, poor growth, lack of tears, frequent lung infections, and difficulty maintaining body temperature. Older infants and young children with familial dysautonomia may hold their breath for prolonged periods of time, which may cause a bluish appearance of the skin or lips (cyanosis) or fainting. This breath-holding behavior usually stops by age 6. Developmental milestones, such as walking and speech, are usually delayed, although some affected individuals show no signs of developmental delay.
Additional signs and symptoms in school-age children include bed wetting, episodes of vomiting, reduced sensitivity to temperature changes and pain, poor balance, abnormal curvature of the spine (scoliosis), poor bone quality and increased risk of bone fractures, and kidney and heart problems. Affected individuals also have poor regulation of blood pressure. They may experience a sharp drop in blood pressure upon standing (orthostatic hypotension), which can cause dizziness, blurred vision, or fainting. They can also have episodes of high blood pressure when nervous or excited, or during vomiting incidents. About one-third of children with familial dysautonomia have learning disabilities, such as a short attention span, that require special education classes. By adulthood, affected individuals often have increasing difficulties with balance and walking unaided. Other problems that may appear in adolescence or early adulthood include lung damage due to repeated infections, impaired kidney function, and worsening vision due to the shrinking size (atrophy) of optic nerves, which carry information from the eyes to the brain.
Type 3, familial dysautonomia (FD) or Riley-Day syndrome, is an autosomal recessive disorder seen predominantly in Jews of eastern European descent. Patients present with sensory and autonomic disturbances. Newborns have absent or weak suck reflex, hypotonia and hypothermia. Delayed physical development, poor temperature and motor incoordination are seen in early childhood. Other features include reduced or absent tears, depressed deep tendon reflexes, absent corneal reflex, postural hypotension and relative indifference to pain. Scoliosis is frequent. Intelligence remains normal. Many patients die in infancy and childhood. Lack of flare with intradermal histamine is seen. Histopathology of peripheral nerve shows reduced number of myelinated and non-myelinated axons. The catecholamine endings are absent.
Genes related to Hereditary sensory and autonomic neuropathy Type 3:
Mutations in the IKBKAP gene cause familial dysautonomia.
The IKBKAP gene provides instructions for making a protein called IKK complex-associated protein (IKAP). This protein is found in a variety of cells throughout the body, including brain cells.
Nearly all individuals with familial dysautonomia have two copies of the same IKBKAP gene mutation in each cell. This mutation can disrupt how information in the IKBKAP gene is pieced together to make a blueprint for the production of IKAP protein. As a result of this error, a reduced amount of normal IKAP protein is produced. This mutation behaves inconsistently, however. Some cells produce near normal amounts of the protein, and other cells—particularly brain cells—have very little of the protein. Critical activities in brain cells are probably disrupted by reduced amounts or the absence of IKAP protein, leading to the signs and symptoms of familial dysautonomia.
Type 4, Congenital insensitivity to pain with anhidrosis
Congenital insensitivity to pain with anhidrosis (CIPA), also known as hereditary sensory and autonomic neuropathy type IV (HSAN IV), is characterized by insensitivity to pain, anhidrosis (the inability to sweat), and intellectual disability. The ability to sense all pain (including visceral pain) is absent, resulting in repeated injuries including: oral self-mutilation (biting of tongue, lips, and buccal mucosa); biting of fingertips; bruising, scarring, and infection of the skin; multiple bone fractures (many of which fail to heal properly); and recurrent joint dislocations resulting in joint deformity. Sense of touch, vibration, and position are normal. Anhidrosis predisposes to recurrent febrile episodes that are often the initial manifestation of CIPA. Hypothermia in cold environments also occurs. Intellectual disability of varying degree is observed in most affected individuals; hyperactivity and emotional lability are common.
Hereditary sensory neuropathy type IV (HSN4) is a rare genetic disorder characterized by the loss of sensation (sensory loss), especially in the feet and legs and, less severely, in the hands and forearms. The sensory loss is due to abnormal functioning of small, unmyelinated nerve fibers and portions of the spinal cord that control responses to pain and temperature as well as other involuntary or automatic body processes. Sweating is almost completely absent with this disorder. Intellectual disability is usually present.
Type 4, congenital insensitivity to pain with anhidrosis (CIPA), is an autosomal recessive condition and affected infants present with episodes of hyperthermia unrelated to environmental temperature, anhidrosis and insensitivity to pain. Palmar skin is thickened and charcot joints are commonly present. NCV shows motor and sensory nerve action potentials to be normal. The histopathology of peripheral nerve biopsy reveals absent small unmyelinated fibers and mitochondria are abnormally enlarged.
Management of Hereditary sensory and autonomic neuropathy Type 4:
Treatment of manifestations: Treatment is supportive and is best provided by specialists in pediatrics, orthopedics, dentistry, ophthalmology, and dermatology. For anhidrosis: Monitoring body temperature helps to institute timely measures to prevent/manage hyperthermia or hypothermia. For insensitivity to pain: Modify as much as reasonable a child's activities to prevent injuries. Inability to provide proper immobilization as a treatment for orthopedic injuries often delays healing; additionally, bracing and invasive orthopedic procedures increase the risk for infection. Methods used to prevent injuries to the lips, buccal mucosa, tongue, and teeth include tooth extraction, and/or filing (smoothing) of the sharp incisal edges of teeth, and/or use of a mouth guard. Skin care with moisturizers can help prevent palmar and plantar hyperkeratosis and cracking and secondary risk of infection; neurotrophic keratitis is best treated with routine care for dry eyes, prevention of corneal infection, and daily observation of the ocular surface. Interventions for behavioral, developmental and motor delays, as well as educational and social support for school-age children and adolescents, are recommended.
Prevention of secondary complications: Regular dental examinations and restriction of sweets to prevent dental caries; early treatment of dental caries and periodontal disease to prevent osteomyelitis of the mandible. During and following surgical procedures, potential complications to identify and manage promptly include hyper- or hypothermia and inadequate sedation, which may trigger unexpected movement and result in secondary injuries.
Type 5, Congenital insensitivity to pain with partial anhidrosis
Hereditary sensory and autonomic neuropathy type V (HSAN5) is a condition that primarily affects the sensory nerve cells (sensory neurons), which transmit information about sensations such as pain, temperature, and touch. These sensations are impaired in people with HSAN5.
The signs and symptoms of HSAN5 appear early, usually at birth or during infancy. People with HSAN5 lose the ability to feel pain, heat, and cold. Deep pain perception, the feeling of pain from injuries to bones, ligaments, or muscles, is especially affected in people with HSAN5. Because of the inability to feel deep pain, affected individuals suffer repeated severe injuries such as bone fractures and joint injuries that go unnoticed. Repeated trauma can lead to a condition called Charcot joints, in which the bones and tissue surrounding joints are destroyed.
Type 5, congenital insensitivity to pain with partial anhidrosis,[4] also manifests with congenital insensitivity to pain & anhidrosis. There is a selective absence of small myelinated fibers differentiating it from Type IV (CIPA).
Genes related to Hereditary sensory and autonomic neuropathy Type 5:
Mutations in the NGF gene cause HSAN5. The NGF gene provides instructions for making a protein called nerve growth factor beta (NGFβ
Type 6 -
Hereditary sensory and autonomic neuropathy type 6 (HSAN6) is a severe autosomal recessive disorder characterized by neonatal hypotonia, respiratory and feeding difficulties, lack of psychomotor development, and autonomic abnormalities including labile cardiovascular function, lack of corneal reflexes leading to corneal scarring, areflexia, and absent axonal flare response after intradermal histamine injection.[5]
Genes related to Hereditary sensory and autonomic neuropathy Type 6:
Mutations in the DST (Dystosin) gene are associated HSAN6. In two separate studies of families with presentations of HSAN6, the DST gene was found to be mutated, in one case with a truncating mutation, and in the other a non-sense or missense mutation. In the latter case, the authors proposed that homozygous truncating mutations result in a severe form of HSAN6, with congenital defects and early lethality, while heterozygosity for a truncating or missense mutation maintains enough dystosin expression and function to allow a normal lifespan, albeit with symptoms of HSAN6.[5]
DST is involved in maintaining cytoskeleton integrity and intracellular transport systems, each of which are critical to the normal function of the particularly long neurons found in the peripheral nervous system. It's believed that this disrupts the cell's internal autophagy process, in which toxic proteins and other normally-occurring debris are captured and transported to the cell center for recycling or destruction. In neurons, autophagosomes, are generated at the ends of the neuron to carry the discardable material, and they are then transported toward center. With the intracellular transport system disrupted by a mutation in DST, the autophagosomes are unable to carry away the debris, which builds up at the ends of the neuron, leading to damage.[6]
This section is empty. You can help by adding to it. (December 2017)
Treatment
This section is empty. You can help by adding to it. (May 2022)
Related Research Articles
Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder. The disease presents with progressive stiffness (spasticity) and contraction in the lower limbs. HSP is also known as hereditary spastic paraparesis, familial spastic paraplegia, French settlement disease, Strumpell disease, or Strumpell-Lorrain disease. The symptoms are a result of dysfunction of long axons in the spinal cord. The affected cells are the primary motor neurons; therefore, the disease is an upper motor neuron disease. HSP is not a form of cerebral palsy even though it physically may appear and behave much the same as spastic diplegia. The origin of HSP is different from cerebral palsy. Despite this, some of the same anti-spasticity medications used in spastic cerebral palsy are sometimes used to treat HSP symptoms.
Autonomic neuropathy is a form of polyneuropathy that affects the non-voluntary, non-sensory nervous system, affecting mostly the internal organs such as the bladder muscles, the cardiovascular system, the digestive tract, and the genital organs. These nerves are not under a person's conscious control and function automatically. Autonomic nerve fibers form large collections in the thorax, abdomen, and pelvis outside the spinal cord. They have connections with the spinal cord and ultimately the brain, however. Most commonly autonomic neuropathy is seen in persons with long-standing diabetes mellitus type 1 and 2. In most—but not all—cases, autonomic neuropathy occurs alongside other forms of neuropathy, such as sensory neuropathy.
Diabetic neuropathy is various types of nerve damage associated with diabetes mellitus. Symptoms depend on the site of nerve damage and can include motor changes such as weakness; sensory symptoms such as numbness, tingling, or pain; or autonomic changes such as urinary symptoms. These changes are thought to result from a microvascular injury involving small blood vessels that supply nerves. Relatively common conditions which may be associated with diabetic neuropathy include distal symmetric polyneuropathy; third, fourth, or sixth cranial nerve palsy; mononeuropathy; mononeuropathy multiplex; diabetic amyotrophy; and autonomic neuropathy.
Peripheral neuropathy, often shortened to neuropathy, refers to damage or disease affecting the nerves. Damage to nerves may impair sensation, movement, gland function, and/or organ function depending on which nerves are affected. Neuropathies affecting motor, sensory, or autonomic nerves result in different symptoms. More than one type of nerve may be affected simultaneously. Peripheral neuropathy may be acute or chronic, and may be reversible or permanent.
Congenital insensitivity to pain (CIP), also known as congenital analgesia, is one or more extraordinarily rare conditions in which a person cannot feel physical pain. The conditions described here are separate from the HSAN group of disorders, which have more specific signs and cause. Because feeling physical pain is vital for survival, CIP is an extremely dangerous condition. It is common for people with the condition to die in childhood due to injuries or illnesses going unnoticed. Burn injuries are among the more common injuries.
Congenital insensitivity to pain with anhidrosis (CIPA) is a rare autosomal recessive disorder of the nervous system which prevents the feeling of pain or temperature, and prevents a person from sweating. Cognitive disorders are commonly coincident. CIPA is the fourth type of hereditary sensory and autonomic neuropathy (HSAN), and is also known as HSAN IV.
Familial dysautonomia (FD), also known as Riley-Day syndrome, is a rare, progressive, recessive genetic disorder of the autonomic nervous system that affects the development and survival of sensory, sympathetic, and some parasympathetic neurons in the autonomic and sensory nervous system.
Giant axonal neuropathy is a rare, autosomal recessive neurological disorder that causes disorganization of neurofilaments. Neurofilaments form a structural framework that helps to define the shape and size of neurons and are essential for normal nerve function. A distinguishing feature is its association with kinky, or curly, hair; in such cases it has been called Giant axonal neuropathy with curly hair.
IKBKAP is a human gene encoding the IKAP protein, which is ubiquitously expressed at varying levels in all tissue types, including brain cells. The IKAP protein is thought to participate as a sub-unit in the assembly of a six-protein putative human holo-Elongator complex, which allows for transcriptional elongation by RNA polymerase II. Further evidence has implicated the IKAP protein as being critical in neuronal development, and directs that decreased expression of IKAP in certain cell types is the molecular basis for the severe, neurodevelopmental disorder familial dysautonomia. Other pathways that have been connected to IKAP protein function in a variety of organisms include tRNA modification, cell motility, and cytosolic stress signalling. Homologs of the IKBKAP gene have been identified in multiple other Eukaryotic model organisms. Notable homologs include Elp1 in yeast, Ikbkap in mice, and D-elp1 in fruit flies. The fruit fly homolog (D-elp1) has RNA-dependent RNA polymerase activity and is involved in RNA interference.
Familial amyloid polyneuropathy, also called transthyretin-related hereditary amyloidosis, transthyretin amyloidosis abbreviated also as ATTR, or Corino de Andrade's disease, is an autosomal dominant neurodegenerative disease. It is a form of amyloidosis, and was first identified and described by Portuguese neurologist Mário Corino da Costa Andrade, in 1952. FAP is distinct from senile systemic amyloidosis (SSA), which is not inherited, and which was determined to be the primary cause of death for 70% of supercentenarians who have been autopsied. FAP can be ameliorated by liver transplantation.
Small fiber peripheral neuropathy is a type of peripheral neuropathy that occurs from damage to the small unmyelinated and myelinated peripheral nerve fibers. These fibers, categorized as C fibers and small Aδ fibers, are present in skin, peripheral nerves, and organs. The role of these nerves is to innervate some skin sensations and help control autonomic function. It is estimated that 15–20 million people in the United States have some form of peripheral neuropathy.
Gap junction beta-1 protein (GJB1), also known as connexin 32 (Cx32), is a transmembrane protein that in humans is encoded by the GJB1 gene. Gap junction beta-1 protein is a member of the gap junction connexin family of proteins that regulates and controls the transfer of communication signals across cell membranes, primarily in the liver and peripheral nervous system. However, the protein is expressed in multiple organs, including in oligodendrocytes in the central nervous system.
Hereditary motor and sensory neuropathies (HMSN) is a name sometimes given to a group of different neuropathies which are all characterized by their impact upon both afferent and efferent neural communication. HMSN are characterised by atypical neural development and degradation of neural tissue. The two common forms of HMSN are either hypertrophic demyelinated nerves or complete atrophy of neural tissue. Hypertrophic condition causes neural stiffness and a demyelination of nerves in the peripheral nervous system, and atrophy causes the breakdown of axons and neural cell bodies. In these disorders, a patient experiences progressive muscle atrophy and sensory neuropathy of the extremities.
Hereditary sensory neuropathy, type II also known as HSN2 is a region of a parent protein which in humans is encoded by the WNK1 gene. It is a transcript variant of the WNK1 gene that is selectively expressed in nervous system tissues, and during development. Mutations in this exon of the WNK1 gene have been identified as causative in genetic neuropathy syndromes, and in inherited pain insensitivity.
A neurological disorder is any disorder of the nervous system. Structural, biochemical or electrical abnormalities in the brain, spinal cord or other nerves can result in a range of symptoms. Examples of symptoms include paralysis, muscle weakness, poor coordination, loss of sensation, seizures, confusion, pain, tauopathies, and altered levels of consciousness. There are many recognized neurological disorders, some relatively common, but many rare. They may be assessed by neurological examination, and studied and treated within the specialties of neurology and clinical neuropsychology.
Hereditary neuropathy with liability to pressure palsy (HNPP) is a peripheral neuropathy, a condition that affects the nerves. Pressure on the nerves can cause tingling sensations, numbness, pain, weakness, muscle atrophy and even paralysis of the affected area. In normal individuals, these symptoms disappear quickly, but in sufferers of HNPP even a short period of pressure can cause the symptoms to occur. Palsies can last from minutes or days to weeks or even months.
Hereditary sensory and autonomic neuropathy type I or hereditary sensory neuropathy type I is a group of autosomal dominant inherited neurological diseases that affect the peripheral nervous system particularly on the sensory and autonomic functions. The hallmark of the disease is the marked loss of pain and temperature sensation in the distal parts of the lower limbs. The autonomic disturbances, if present, manifest as sweating abnormalities.
PR domain zinc finger protein 12 is a protein that in humans is encoded by the PRDM12 gene. This gene is normally switched on during the development of pain-sensing nerve cells. People with homozygous mutations of the PRDM12 gene experience congenital insensitivity to pain (CIP). PRMD12 is a part of a larger domain that mediate histone methyltransferases. Enzymes target gene promoters in order to control gene expression.
Marsili syndrome is an extremely rare genetic disorder which is characterized by symptoms similar to those reported on individuals with congenital insensitivity to pain with anhidrosis. It can be fatal if it goes unnoticed/undiagnosed.
↑ Minde J, Toolanen G, Andersson T, etal. (December 2004). "Familial insensitivity to pain (HSAN V) and a mutation in the NGFB gene. A neurophysiological and pathological study". Muscle Nerve. 30 (6): 752–760. doi:10.1002/mus.20172. PMID15468048. S2CID23764234.
↑ Henry Houlden; R. H. M. King; A. Hashemi-Nejad; etal. (April 2001). "A novel TRK A (NTRK1) mutation associated with hereditary sensory and autonomic neuropathy type V". Annals of Neurology. 49 (4): 521–525. doi:10.1002/ana.103. PMID11310631. S2CID7888893.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.