The Vienna Ab initio Simulation Package, better known as VASP, is a package written primarily in Fortran for performing ab initio quantum mechanical calculations using either Vanderbilt pseudopotentials, or the projector augmented wave method, and a plane wave basis set. The basic methodology is density functional theory (DFT), but the code also allows use of post-DFT corrections such as hybrid functionals mixing DFT and Hartree–Fock exchange, many-body perturbation theory and dynamical electronic correlations within the random phase approximation (RPA) and MP2.
Atomistix Virtual NanoLab (VNL) is a commercial point-and-click software for simulation and analysis of physical and chemical properties of nanoscale devices. Virtual NanoLab is developed and sold commercially by QuantumWise A/S. QuantumWise was then acquired by Synopsys in 2017.
Car–Parrinello molecular dynamics or CPMD refers to either a method used in molecular dynamics or the computational chemistry software package used to implement this method.
Atomistix ToolKit (ATK) is a commercial software for atomic-scale modeling and simulation of nanosystems. The software was originally developed by Atomistix A/S, and was later acquired by QuantumWise following the Atomistix bankruptcy. QuantumWise was then acquired by Synopsys in 2017.
NanoLanguage is a scripting interface built on top of the interpreted programming language Python, and is primarily intended for simulation of physical and chemical properties of nanoscale systems.
This is a list of computer programs that are predominantly used for molecular mechanics calculations.
Materials Studio is software for simulating and modeling materials. It is developed and distributed by BIOVIA, a firm specializing in research software for computational chemistry, bioinformatics, cheminformatics, molecular dynamics simulation, and quantum mechanics.
The following outline is provided as an overview of and topical guide to nanotechnology:
Nanomechanics is a branch of nanoscience studying fundamental mechanical properties of physical systems at the nanometer scale. Nanomechanics has emerged on the crossroads of biophysics, classical mechanics, solid-state physics, statistical mechanics, materials science, and quantum chemistry. As an area of nanoscience, nanomechanics provides a scientific foundation of nanotechnology.

In condensed-matter physics, a collision cascade is a set of nearby adjacent energetic collisions of atoms induced by an energetic particle in a solid or liquid.

Molecular modeling on GPU is the technique of using a graphics processing unit (GPU) for molecular simulations.

Ascalaph Designer is a computer program for general purpose molecular modelling for molecular design and simulations. It provides a graphical environment for the common programs of quantum and classical molecular modelling ORCA, NWChem, Firefly, CP2K and MDynaMix . The molecular mechanics calculations cover model building, energy optimizations and molecular dynamics. Firefly covers a wide range of quantum chemistry methods. Ascalaph Designer is free and open-source software, released under the GNU General Public License, version 2 (GPLv2).
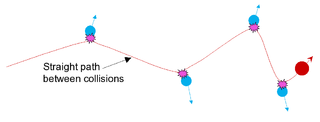
In condensed-matter physics, the binary collision approximation (BCA) is a heuristic used to more efficiently simulate the penetration depth and defect production by energetic ions in solids. In the method, the ion is approximated to travel through a material by experiencing a sequence of independent binary collisions with sample atoms (nuclei). Between the collisions, the ion is assumed to travel in a straight path, experiencing electronic stopping power, but losing no energy in collisions with nuclei.

Ninithi is free and open source modelling software that can be used to visualize and analyze carbon materials used in nanotechnology. Users of ninithi can visualize the 3D molecular geometries of graphene/nano-ribbons, carbon nanotubes and fullerenes. Ninithi also provides features to simulate the electronic band structures of graphene and carbon nanotubes. The software was developed by Lanka Software Foundation, in Sri Lanka and released in 2010 under the GPL licence. Ninithi is written in the Java programming language and available for both Microsoft Windows and Linux platforms.

Lee Young-hee is a South Korean physicist. He is a distinguished professor in physics and energy science at Sungkyunkwan University as a SKKU fellow. He is also director of the Center for Integrated Nanostructure Physics in the Institute for Basic Science (IBS). He has been a Clarivate Analytics Highly Cited Researcher in the cross-field category in 2018–2023.
Alan T. Charlie Johnson is an American physicist, a professor in physics and astronomy at the University of Pennsylvania, and was the director of the Nano/Bio Interface Center at the University of Pennsylvania (2014-2017).

SAMSON is a computer software platform for molecular design being developed by OneAngstrom and previously by the NANO-D group at the French Institute for Research in Computer Science and Automation (INRIA).

Oleg V. Prezhdo is a Ukrainian–American physical chemist whose research focuses on non-adiabatic molecular dynamics and time-dependent density functional theory (TDDFT). His research interests range from fundamental aspects of semi-classical and quantum-classical physics to excitation dynamics in condensed matter and biological systems. His research group focuses on the development of new theoretical models and computational tools aimed at understanding chemical reactivity and energy transfer at a molecular level in complex condensed phase environment. Since 2014, he is a professor of chemistry and of physics & astronomy at the University of Southern California.
MBN Explorer is a software package for molecular dynamics simulations, structure optimization and kinetic Monte Carlo simulations. It is designed for multiscale computational analysis of structure and dynamics of atomic clusters and nanoparticles, biomolecules and nanosystems, nanostructured materials, different states of matter and various interfaces. The software has been developed by MBN Research Center.
In the context of chemistry and molecular modelling, the Interface force field (IFF) is a force field for classical molecular simulations of atoms, molecules, and assemblies up to the large nanometer scale, covering compounds from across the periodic table. It employs a consistent classical Hamiltonian energy function for metals, oxides, and organic compounds, linking biomolecular and materials simulation platforms into a single platform. The reliability is often higher than that of density functional theory calculations at more than a million times lower computational cost. IFF includes a physical-chemical interpretation for all parameters as well as a surface model database that covers different cleavage planes and surface chemistry of included compounds. The Interface Force Field is compatible with force fields for the simulation of primarily organic compounds and can be used with common molecular dynamics and Monte Carlo codes. Structures and energies of included chemical elements and compounds are rigorously validated and property predictions are up to a factor of 100 more accurate relative to earlier models.
