Clinical trials are prospective biomedical or behavioral research studies on human participants designed to answer specific questions about biomedical or behavioral interventions, including new treatments and known interventions that warrant further study and comparison. Clinical trials generate data on dosage, safety and efficacy. They are conducted only after they have received health authority/ethics committee approval in the country where approval of the therapy is sought. These authorities are responsible for vetting the risk/benefit ratio of the trial—their approval does not mean the therapy is 'safe' or effective, only that the trial may be conducted.

A vaccine trial is a clinical trial that aims at establishing the safety and efficacy of a vaccine prior to it being licensed.
Pharmacovigilance, also known as drug safety, is the pharmaceutical science relating to the "collection, detection, assessment, monitoring, and prevention" of adverse effects with pharmaceutical products. The etymological roots for the word "pharmacovigilance" are: pharmakon and vigilare. As such, pharmacovigilance heavily focuses on adverse drug reactions (ADR), which are defined as any response to a drug which is noxious and unintended, including lack of efficacy. Medication errors such as overdose, and misuse and abuse of a drug as well as drug exposure during pregnancy and breastfeeding, are also of interest, even without an adverse event, because they may result in an adverse drug reaction.
An adverse event (AE) is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product.
A serious adverse event (SAE) in human drug trials is defined as any untoward medical occurrence that at any dose
- Results in death
- Is life-threatening
- Requires inpatient hospitalization or causes prolongation of existing hospitalization
- Results in persistent or significant disability/incapacity
- May have caused a congenital anomaly/birth defect
- Requires intervention to prevent permanent impairment or damage
Good clinical practice (GCP) is an international quality standard, which governments can then transpose into regulations for clinical trials involving human subjects. GCP follows the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), and enforces tight guidelines on ethical aspects of clinical research.
Clinical research is a branch of healthcare science that determines the safety and effectiveness (efficacy) of medications, devices, diagnostic products and treatment regimens intended for human use. These may be used for prevention, treatment, diagnosis or for relieving symptoms of a disease. Clinical research is different from clinical practice. In clinical practice established treatments are used, while in clinical research evidence is collected to establish a treatment.
A data monitoring committee (DMC) – sometimes called a data and safety monitoring board (DSMB) – is an independent group of experts who monitor patient safety and treatment efficacy data while a clinical trial is ongoing.
Postmarketing surveillance (PMS), also known as post market surveillance, is the practice of monitoring the safety of a pharmaceutical drug or medical device after it has been released on the market and is an important part of the science of pharmacovigilance. Since drugs and medical devices are approved on the basis of clinical trials, which involve relatively small numbers of people who have been selected for this purpose – meaning that they normally do not have other medical conditions which may exist in the general population – postmarketing surveillance can further refine, or confirm or deny, the safety of a drug or device after it is used in the general population by large numbers of people who have a wide variety of medical conditions.
In drug development and medical device development the Investigator's Brochure (IB) is a comprehensive document summarizing the body of information about an investigational product obtained during a drug trial. The IB is a document of critical importance throughout the drug development process and is updated with new information as it becomes available. The purpose of the IB is to compile data relevant to studies of the IP in human subjects gathered during preclinical and other clinical trials.
A Clinical Research Coordinator (CRC) is a person responsible for conducting clinical trials using good clinical practice (GCP) under the auspices of a Principal Investigator (PI).

President of the United States George W. Bush signed the Food and Drug Administration Amendments Act of 2007 (FDAAA) on September 27, 2007. This law reviewed, expanded, and reaffirmed several existing pieces of legislation regulating the FDA. These changes allow the FDA to perform more comprehensive reviews of potential new drugs and devices. It was sponsored by Reps. Joe Barton and Frank Pallone and passed unanimously by the Senate.
Safety pharmacology is a branch of pharmacology specialising in detecting and investigating potential undesirable pharmacodynamic effects of new chemical entities (NCEs) on physiological functions in relation to exposure in the therapeutic range and above.
A glossary of terms used in clinical research.
The following outline is provided as an overview of and topical guide to clinical research:
Patient recruitment includes a variety of services—typically performed by a Patient Recruitment Service Provider—to increase enrollment into clinical trials. Presently, the patient recruitment industry is claimed to total $19 billion per year.
The phases of clinical research are the stages in which scientists conduct experiments with a health intervention to obtain sufficient evidence for a process considered effective as a medical treatment. For drug development, the clinical phases start with testing for drug safety in a few human subjects, then expand to many study participants to determine if the treatment is effective. Clinical research is conducted on drug candidates, vaccine candidates, new medical devices, and new diagnostic assays.
An independent safety officer (ISO) is a clinician or researcher who is independent of the clinical study team and helps to monitor a clinical trial for research participant (patient) safety, adverse events, trial progress, and data quality. An ISO has relevant experience with clinical trials as well as with the patient population and intervention being studied. Clinical trials using an ISO are usually smaller, single-site trials with a moderate to minimal risk intervention.
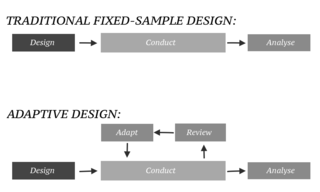
In an adaptive design of a clinical trial, the parameters and conduct of the trial for a candidate drug or vaccine may be changed based on an interim analysis. Adaptive design typically involves advanced statistics to interpret a clinical trial endpoint. This is in contrast to traditional single-arm clinical trials or randomized clinical trials (RCTs) that are static in their protocol and do not modify any parameters until the trial is completed. The adaptation process takes place at certain points in the trial, prescribed in the trial protocol. Importantly, this trial protocol is set before the trial begins with the adaptation schedule and processes specified. Adaptions may include modifications to: dosage, sample size, drug undergoing trial, patient selection criteria and/or "cocktail" mix. The PANDA provides not only a summary of different adaptive designs, but also comprehensive information on adaptive design planning, conduct, analysis and reporting.