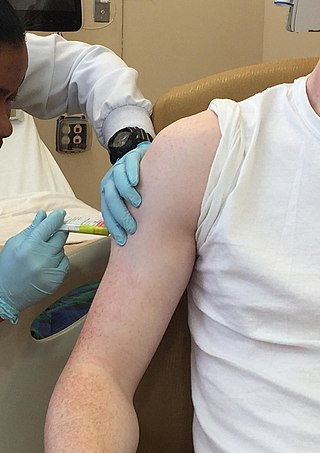
Clinical trials are prospective biomedical or behavioral research studies on human participants designed to answer specific questions about biomedical or behavioral interventions, including new treatments and known interventions that warrant further study and comparison. Clinical trials generate data on dosage, safety and efficacy. They are conducted only after they have received health authority/ethics committee approval in the country where approval of the therapy is sought. These authorities are responsible for vetting the risk/benefit ratio of the trial—their approval does not mean the therapy is 'safe' or effective, only that the trial may be conducted.
A medical error is a preventable adverse effect of care ("iatrogenesis"), whether or not it is evident or harmful to the patient. This might include an inaccurate or incomplete diagnosis or treatment of a disease, injury, syndrome, behavior, infection, or other ailment.
Pharmacovigilance, also known as drug safety, is the pharmaceutical science relating to the "collection, detection, assessment, monitoring, and prevention" of adverse effects with pharmaceutical products. The etymological roots for the word "pharmacovigilance" are: pharmakon and vigilare. As such, pharmacovigilance heavily focuses on adverse drug reactions (ADR), which are defined as any response to a drug which is noxious and unintended, including lack of efficacy. Medication errors such as overdose, and misuse and abuse of a drug as well as drug exposure during pregnancy and breastfeeding, are also of interest, even without an adverse event, because they may result in an adverse drug reaction.
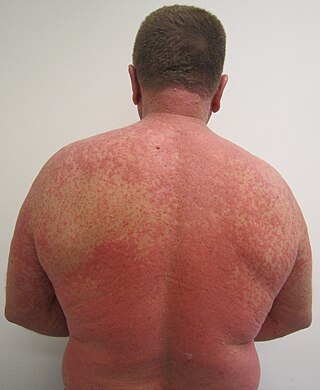
An adverse drug reaction (ADR) is a harmful, unintended result caused by taking medication. ADRs may occur following a single dose or prolonged administration of a drug or may result from the combination of two or more drugs. The meaning of this term differs from the term "side effect" because side effects can be beneficial as well as detrimental. The study of ADRs is the concern of the field known as pharmacovigilance. An adverse event (AE) refers to any unexpected and inappropriate occurrence at the time a drug is used, whether or not the event is associated with the administration of the drug. An ADR is a special type of AE in which a causative relationship can be shown. ADRs are only one type of medication-related harm. Another type of medication-related harm type includes not taking prescribed medications, which is also known as non-adherence. Non-adherence to medications can lead to death and other negative outcomes. Adverse drug reactions require the use of a medication.

The Food and Drug Administration's (FDA) New Drug Application (NDA) is the vehicle in the United States through which drug sponsors formally propose that the FDA approve a new pharmaceutical for sale and marketing. Some 30% or less of initial drug candidates proceed through the entire multi-year process of drug development, concluding with an approved NDA, if successful.
An adverse effect is an undesired harmful effect resulting from a medication or other intervention, such as surgery. An adverse effect may be termed a "side effect", when judged to be secondary to a main or therapeutic effect. The term complication is similar to adverse effect, but the latter is typically used in pharmacological contexts, or when the negative effect is expected or common. If the negative effect results from an unsuitable or incorrect dosage or procedure, this is called a medical error and not an adverse effect. Adverse effects are sometimes referred to as "iatrogenic" because they are generated by a physician/treatment. Some adverse effects occur only when starting, increasing or discontinuing a treatment. Adverse effects can also be caused by placebo treatments . Using a drug or other medical intervention which is contraindicated may increase the risk of adverse effects. Adverse effects may cause complications of a disease or procedure and negatively affect its prognosis. They may also lead to non-compliance with a treatment regimen. Adverse effects of medical treatment resulted in 142,000 deaths in 2013 up from 94,000 deaths in 1990 globally.
The United States Food and Drug Administration's Investigational New Drug (IND) program is the means by which a pharmaceutical company obtains permission to start human clinical trials and to ship an experimental drug across state lines before a marketing application for the drug has been approved. Regulations are primarily at 21 CFR 312. Similar procedures are followed in the European Union, Japan, and Canada.
Clinical monitoring is the oversight and administrative efforts that monitor a participant's health and efficacy of the treatment during a clinical trial. Both independent and government-run grant-funding agencies, such as the National Institutes of Health (NIH) and the World Health Organization (WHO), require data and safety monitoring protocols for Phase I and II clinical trials conforming to their standards.
An adverse event (AE) is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign, symptom, or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product.

Varenicline, sold under the brand names Chantix and Champix among others, is a medication used for smoking cessation and for the treatment of dry eye disease. It is a nicotinic receptor partial agonist and a cholinergic agonist. When activated, this receptor releases dopamine in the nucleus accumbens, the brain's reward center, thereby reducing cravings and withdrawal symptoms associated with smoking cessation.
In drug development and medical device development the Investigator's Brochure (IB) is a comprehensive document summarizing the body of information about an investigational product obtained during a drug trial. The IB is a document of critical importance throughout the drug development process and is updated with new information as it becomes available. The purpose of the IB is to compile data relevant to studies of the IP in human subjects gathered during preclinical and other clinical trials.
A clinical prediction rule or clinical probability assessment specifies how to use medical signs, symptoms, and other findings to estimate the probability of a specific disease or clinical outcome.
A Clinical Research Coordinator (CRC) is a person responsible for conducting clinical trials using good clinical practice (GCP) under the auspices of a Principal Investigator (PI).
Safety pharmacology is a branch of pharmacology specialising in detecting and investigating potential undesirable pharmacodynamic effects of new chemical entities (NCEs) on physiological functions in relation to exposure in the therapeutic range and above.
A seeding trial or marketing trial is a form of marketing, conducted in the name of research, designed to target product sampling towards selected consumers. In the marketing research field, seeding is the process of allocating marketing to specific customers, or groups of customers, in order to stimulate the internal dynamics of the market, and enhance the diffusion process. In medicine, seeding trials are clinical trials or research studies in which the primary objective is to introduce the concept of a particular medical intervention—such as a pharmaceutical drug or medical device—to physicians, rather than to test a scientific hypothesis.
A glossary of terms used in clinical research.
The following outline is provided as an overview of and topical guide to clinical research:
The Drug Industry Documents Archive (DIDA) is a digital archive of pharmaceutical industry documents created and maintained by the University of California, San Francisco, Library and Center for Knowledge Management. DIDA is a part of the larger UCSF Industry Documents Library which includes the Truth Tobacco Industry Documents. The archive contains documents about pharmaceutical industry clinical trials, publication of study results, pricing, marketing, relations with physicians and drug company involvement in continuing medical education.
The FDA Adverse Event Reporting System is a computerized information database designed to support the U.S. Food and Drug Administration's (FDA) postmarketing safety surveillance program for all approved drug and therapeutic biologic products. The FDA uses FAERS to monitor for new adverse events and medication errors that might occur with these products. It is a system that measures occasional harms from medications to ascertain whether the risk–benefit ratio is high enough to justify continued use of any particular drug and to identify correctable and preventable problems in health care delivery. The system interacts with several related systems including MedWatch and the Vaccine Adverse Event Reporting System. FAERS replaced legacy AERS system in Sep 2012.