
Methylmalonic acidemia, also called methylmalonic aciduria, is an autosomal recessive metabolic disorder that disrupts normal amino acid metabolism. It is a classical type of organic acidemia. The result of this condition is the inability to properly digest specific fats and proteins, which in turn leads to a buildup of a toxic level of methylmalonic acid in the blood.

Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to brain injury and death. It may be primary or secondary.
Online Mendelian Inheritance in Man (OMIM) is a continuously updated catalog of human genes and genetic disorders and traits, with a particular focus on the gene-phenotype relationship. As of 28 June 2019, approximately 9,000 of the over 25,000 entries in OMIM represented phenotypes; the rest represented genes, many of which were related to known phenotypes.

Zellweger syndrome is a rare congenital disorder characterized by the reduction or absence of functional peroxisomes in the cells of an individual. It is one of a family of disorders called Zellweger spectrum disorders which are leukodystrophies. Zellweger syndrome is named after Hans Zellweger (1909–1990), a Swiss-American pediatrician, a professor of pediatrics and genetics at the University of Iowa who researched this disorder.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.
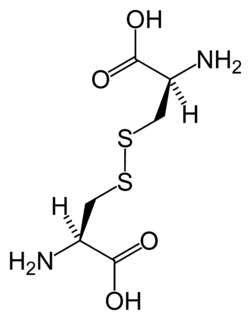
Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.

Salla disease (SD) is an autosomal recessive lysosomal storage disease characterized by early physical impairment and intellectual disability. It was first described in 1979, after Salla, a municipality in Finnish Lapland. Salla disease is one of 40 Finnish heritage diseases and affects approximately 130 individuals, mainly from Finland and Sweden.

Methylmalonyl-CoA mutase is a mitochondrial homodimer apoenzyme that focuses on the catalysis of methylmalonyl CoA to succinyl CoA. The enzyme is bound to adenosylcobalamin, a hormonal derivative of vitamin B12 in order to function. Methylmalonyl-CoA mutase deficiency is caused by genetic defect in the MUT gene responsible for encoding the enzyme. Deficiency in this enzyme accounts for 60% of the cases of methylmalonic acidemia.
Refsum disease is an autosomal recessive neurological disease that results in the over-accumulation of phytanic acid in cells and tissues. It is one of several disorders named after Norwegian neurologist Sigvald Bernhard Refsum (1907–1991). Refsum disease typically is adolescent onset and is diagnosed by above average levels of phytanic acid. Humans obtain the necessary phytanic acid primarily through diet. It is still unclear what function phytanic acid plays physiologically in humans, but has been found to regulate fatty acid metabolism in the liver of mice.

Cutis laxa or pachydermatocele is a group of rare connective tissue disorders in which the skin becomes inelastic and hangs loosely in folds.
Peroxisomal disorders represent a class of medical conditions caused by defects in peroxisome functions. This may be due to defects in single enzymes important for peroxisome function or in peroxins, proteins encoded by PEX genes that are critical for normal peroxisome assembly and biogenesis.
Infantile Refsum disease (IRD) is a rare autosomal recessive congenital peroxisomal biogenesis disorder within the Zellweger spectrum. These are disorders of the peroxisomes that are clinically similar to Zellweger syndrome and associated with mutations in the PEX family of genes. IRD is associated with deficient phytanic acid catabolism, as is adult Refsum disease, but they are different disorders that should not be confused.

Peroxisome biogenesis factor 1, also known as PEX1, is a protein which in humans is encoded by the PEX1 gene.

Peroxisomal biogenesis factor 2 is a protein that in humans is encoded by the PEX2 gene.

Peroxisome assembly factor 2 is a protein that in humans is encoded by the PEX6 gene. PEX6 is an AAA ATPase that localizes to the peroxisome. PEX6 forms a hexamer with PEX1 and is recruited to the membrane by PEX26.
Trichothiodystrophy (TTD) is an autosomal recessive inherited disorder characterised by brittle hair and intellectual impairment. The word breaks down into tricho – "hair", thio – "sulphur", and dystrophy – "wasting away" or literally "bad nourishment". TTD is associated with a range of symptoms connected with organs of the ectoderm and neuroectoderm. TTD may be subclassified into four syndromes: Approximately half of all patients with trichothiodystrophy have photosensitivity, which divides the classification into syndromes with or without photosensitivity; BIDS and PBIDS, and IBIDS and PIBIDS. Modern covering usage is TTD-P (photosensitive), and TTD.

Worth syndrome, also known as benign form of Worth hyperostosis corticalis generalisata with torus platinus, autosomal dominant osteosclerosis, autosomal dominant endosteal hyperostosis or Worth disease, is a rare autosomal dominant congenital disorder that is caused by a mutation in the LRP5 gene. It is characterized by increased bone density and benign bony structures on the palate.
Nasodigitoacoustic syndrome, also called Keipert syndrome, is a rare congenital syndrome first described by J.A. Keipert and colleagues in 1973. The syndrome is characterized by a misshaped nose, broad thumbs and halluces, brachydactyly, sensorineural hearing loss, facial features such as hypertelorism, and developmental delay.
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders caused by genetic mutations and characterised by progressive atrophy of various parts of the brain such as the cerebellum or brainstem. Where known, these disorders are inherited in an autosomal recessive fashion. There is no known cure for PCH.
Zellweger spectrum disorders are a group of rare disorders that create the same disease process. The subdivisions of this spectrum are hyperpipecolic acidemia, Infantile Refsum disease, neonatal adrenoleukodystrophy (NALD), and Zellweger syndrome. It can also be referred to as Peroxisomal Biogenesis Disorders, Zellweger Syndrome Spectrum, NALD, Cerebrohepatorenal Syndrome, and ZSS. It can affect many body organs, including the kidneys, eyes, and hearing. It is named after Hans Zellweger.
