The aldol reaction is a reaction that combines two carbonyl compounds to form a new β-hydroxy carbonyl compound.
Reductive amination is a form of amination that involves the conversion of a carbonyl group to an amine via an intermediate imine. The carbonyl group is most commonly a ketone or an aldehyde. It is a common method to make amines and is widely used in green chemistry since it can be done catalytically in one-pot under mild conditions. In biochemistry, dehydrogenase enzymes use reductive amination to produce the amino acid, glutamate. Additionally, there is ongoing research on alternative synthesis mechanisms which various metal catalysts which require more mild reaction conditions, allowing the reaction to be less energy taxing. Investigation into biocatalysts, such as EnelRED, have allowed for the reduction of chiral amines which is an important factor in pharmaceutical synthesis.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of taxol. Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.

The Johnson–Corey–Chaykovsky reaction is a chemical reaction used in organic chemistry for the synthesis of epoxides, aziridines, and cyclopropanes. It was discovered in 1961 by A. William Johnson and developed significantly by E. J. Corey and Michael Chaykovsky. The reaction involves addition of a sulfur ylide to a ketone, aldehyde, imine, or enone to produce the corresponding 3-membered ring. The reaction is diastereoselective favoring trans substitution in the product regardless of the initial stereochemistry. The synthesis of epoxides via this method serves as an important retrosynthetic alternative to the traditional epoxidation reactions of olefins.

The Petasis reaction is the multi-component reaction of an amine, a carbonyl, and a vinyl- or aryl-boronic acid to form substituted amines.
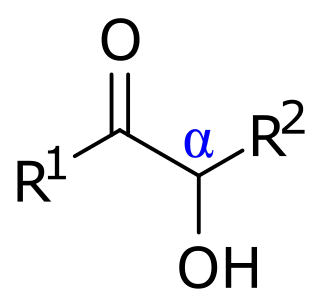
In organic chemistry, acyloins or α-hydroxy ketones are a class of organic compounds of the general form R−C(=O)−CR'(OH)−R", composed of a hydroxy group adjacent to a ketone group. The name acyloin is derived from the fact that they are formally derived from reductive coupling of carboxylic acyl groups. They are one of the two main classes of hydroxy ketones, distinguished by the position of the hydroxy group relative to the ketone; in this form, the hydroxy is on the alpha carbon, explaining the secondary name of α-hydroxy ketone.
In chemistry, transfer hydrogenation is a chemical reaction involving the addition of hydrogen to a compound from a source other than molecular H2. It is applied in laboratory and industrial organic synthesis to saturate organic compounds and reduce ketones to alcohols, and imines to amines. It avoids the need for high-pressure molecular H2 used in conventional hydrogenation. Transfer hydrogenation usually occurs at mild temperature and pressure conditions using organic or organometallic catalysts, many of which are chiral, allowing efficient asymmetric synthesis. It uses hydrogen donor compounds such as formic acid, isopropanol or dihydroanthracene, dehydrogenating them to CO2, acetone, or anthracene respectively. Often, the donor molecules also function as solvents for the reaction. A large scale application of transfer hydrogenation is coal liquefaction using "donor solvents" such as tetralin.
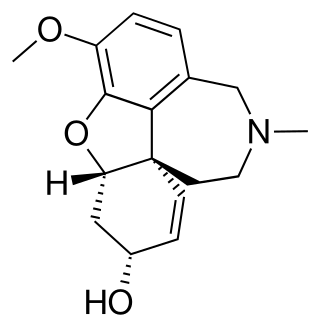
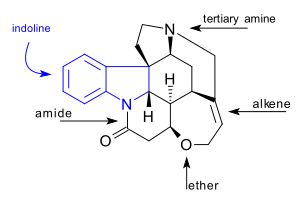
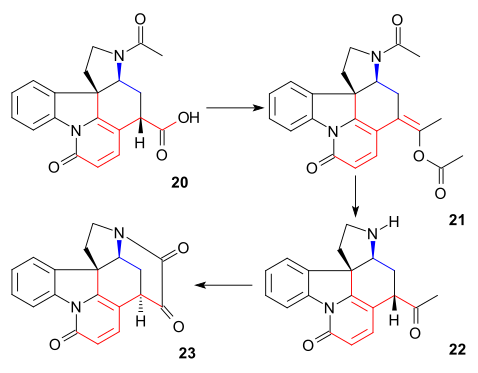
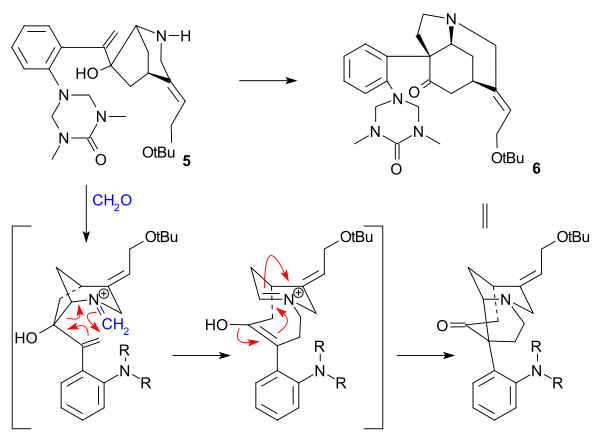
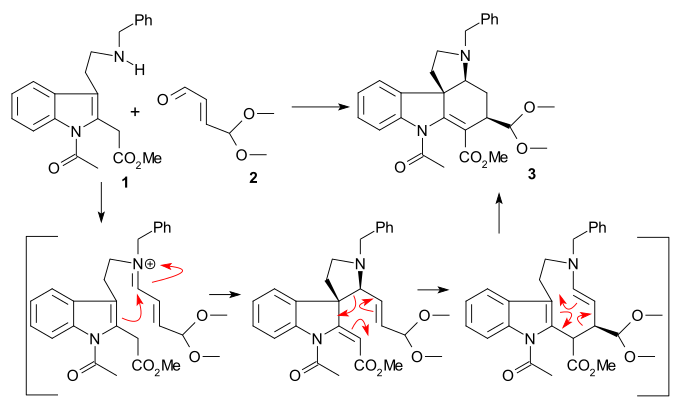
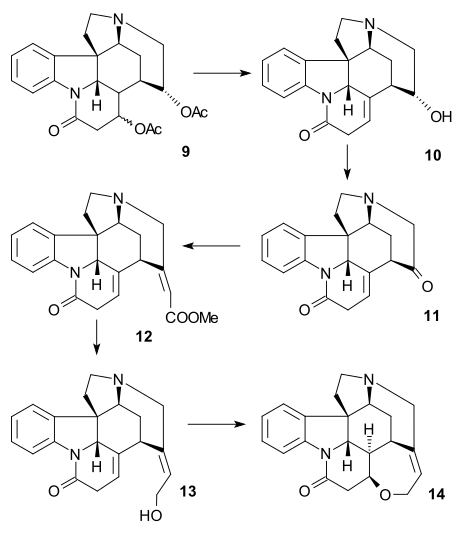
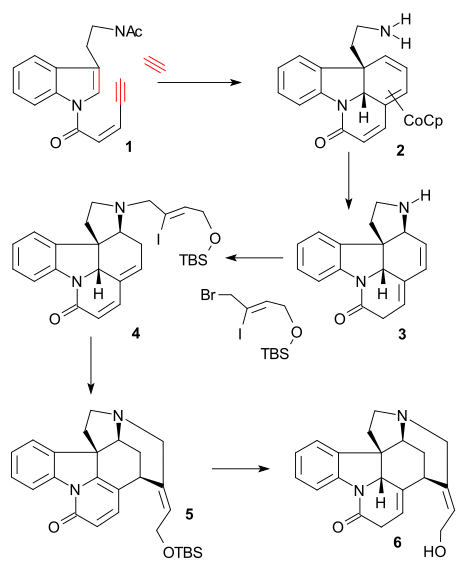
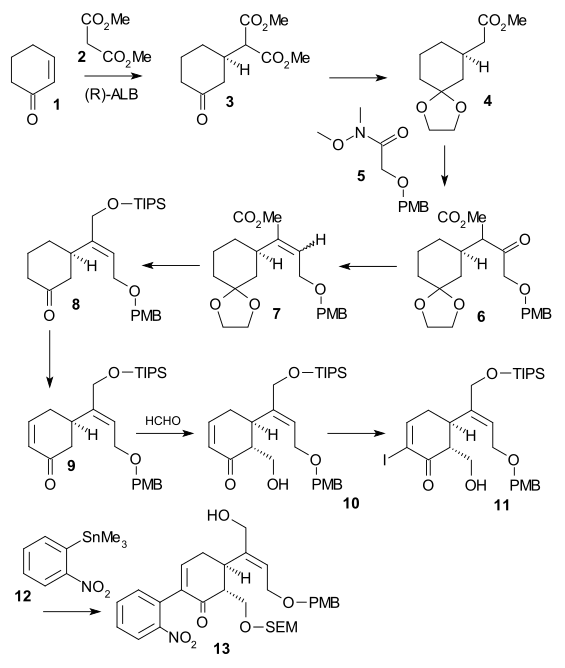
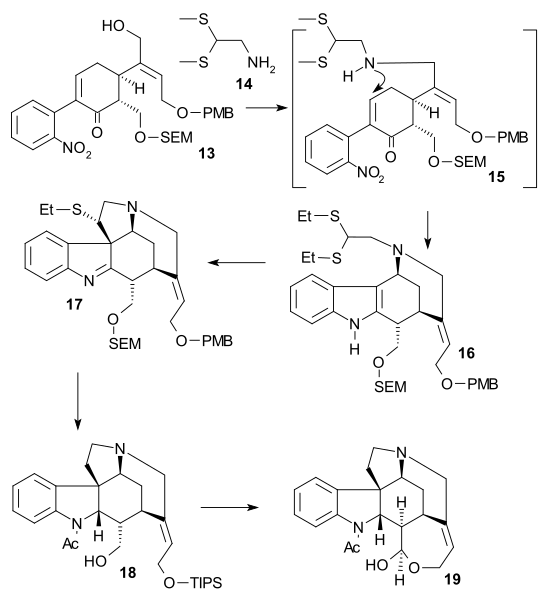
The article concerns the total synthesis of galanthamine, a drug used for the treatment of mild to moderate Alzheimer's disease.
In organic chemistry, kinetic resolution is a means of differentiating two enantiomers in a racemic mixture. In kinetic resolution, two enantiomers react with different reaction rates in a chemical reaction with a chiral catalyst or reagent, resulting in an enantioenriched sample of the less reactive enantiomer. As opposed to chiral resolution, kinetic resolution does not rely on different physical properties of diastereomeric products, but rather on the different chemical properties of the racemic starting materials. The enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction. Kinetic resolution relies upon differences in reactivity between enantiomers or enantiomeric complexes.

In organic chemistry, organocatalysis is a form of catalysis in which the rate of a chemical reaction is increased by an organic catalyst. This "organocatalyst" consists of carbon, hydrogen, sulfur and other nonmetal elements found in organic compounds. Because of their similarity in composition and description, they are often mistaken as a misnomer for enzymes due to their comparable effects on reaction rates and forms of catalysis involved.
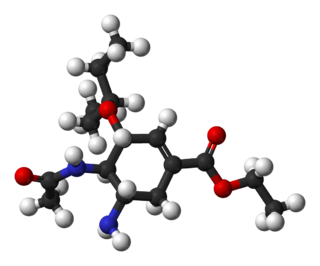
Oseltamivir total synthesis concerns the total synthesis of the antiinfluenza drug oseltamivir marketed by Hoffmann-La Roche under the trade name Tamiflu. Its commercial production starts from the biomolecule shikimic acid harvested from Chinese star anise and from recombinant E. coli. Control of stereochemistry is important: the molecule has three stereocenters and the sought-after isomer is only 1 of 8 stereoisomers.
The total synthesis of quinine, a naturally-occurring antimalarial drug, was developed over a 150-year period. The development of synthetic quinine is considered a milestone in organic chemistry although it has never been produced industrially as a substitute for natural occurring quinine. The subject has also been attended with some controversy: Gilbert Stork published the first stereoselective total synthesis of quinine in 2001, meanwhile shedding doubt on the earlier claim by Robert Burns Woodward and William Doering in 1944, claiming that the final steps required to convert their last synthetic intermediate, quinotoxine, into quinine would not have worked had Woodward and Doering attempted to perform the experiment. A 2001 editorial published in Chemical & Engineering News sided with Stork, but the controversy was eventually laid to rest once and for all when Williams and coworkers successfully repeated Woodward's proposed conversion of quinotoxine to quinine in 2007.

Diisopinocampheylborane is an organoborane that is useful for asymmetric synthesis. This colourless solid is the precursor to a range of related reagents. The compound was reported in 1961 by Zweifel and Brown in a pioneering demonstration of asymmetric synthesis using boranes. The reagent is mainly used for the synthesis of chiral secondary alcohols. The reagent is often depicted as a monomer but like most hydroboranes, it is dimeric with B-H-B bridges.

Borane dimethylsulfide (BMS) is a chemical compound with the chemical formula BH3·S(CH3)2. It is an adduct between borane molecule and dimethyl sulfide molecule. It is a complexed borane reagent that is used for hydroborations and reductions. The advantages of BMS over other borane reagents, such as borane-tetrahydrofuran, are its increased stability and higher solubility. BMS is commercially available at much higher concentrations than its tetrahydrofuran counterpart and does not require sodium borohydride as a stabilizer, which could result in undesired side reactions. In contrast, BH3·THF requires sodium borohydride to inhibit reduction of THF to tributyl borate. BMS is soluble in most aprotic solvents.
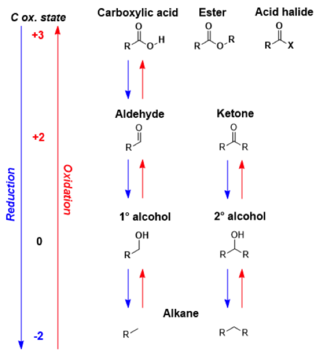
In organic chemistry, carbonyl reduction is the conversion of any carbonyl group, usually to an alcohol. It is a common transformation that is practiced in many ways. Ketones, aldehydes, carboxylic acids, esters, amides, and acid halides - some of the most pervasive functional group]]s, -comprise carbonyl compounds. Carboxylic acids, esters, and acid halides can be reduced to either aldehydes or a step further to primary alcohols, depending on the strength of the reducing agent. Aldehydes and ketones can be reduced respectively to primary and secondary alcohols. In deoxygenation, the alcohol group can be further reduced and removed altogether by replacement with H.
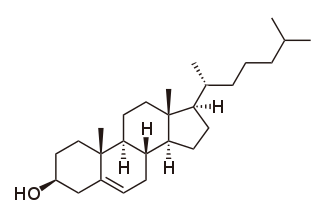
Cholesterol total synthesis in chemistry describes the total synthesis of the complex biomolecule cholesterol and is considered a great scientific achievement. The research group of Robert Robinson with John Cornforth published their synthesis in 1951 and that of Robert Burns Woodward with Franz Sondheimer in 1952. Both groups competed for the first publication since 1950 with Robinson having started in 1932 and Woodward in 1949. According to historian Greg Mulheirn the Robinson effort was hampered by his micromanagement style of leadership and the Woodward effort was greatly facilitated by his good relationships with chemical industry. Around 1949 steroids like cortisone were produced from natural resources but expensive. Chemical companies Merck & Co. and Monsanto saw commercial opportunities for steroid synthesis and not only funded Woodward but also provided him with large quantities of certain chemical intermediates from pilot plants. Hard work also helped the Woodward effort: one of the intermediate compounds was named Christmasterone as it was synthesized on Christmas Day 1950 by Sondheimer.
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.
The asymmetric addition of alkynylzinc compounds to aldehydes is an example of a Nef synthesis, a chemical reaction whereby a chiral propargyl alcohol is prepared from a terminal alkyne and an aldehyde. This alkynylation reaction is enantioselective and involves an alkynylzinc reagent rather than the sodium acetylide used by John Ulric Nef in his 1899 report of the synthetic approach. Propargyl alcohols are versatile precursors for the chirally-selective synthesis of natural products and pharmaceutical agents, making this asymmetric addition reaction of alkynylzinc compounds useful. For example, Erick Carreira used this approach in a total synthesis of the marine natural product leucascandrolide A, a bioactive metabolite of the calcareous sponge Leucascandra caveolata with cytotoxic and antifungal properties isolated in 1996.
Shiina esterification is an organic chemical reaction that synthesizes carboxylic esters from nearly equal amounts of carboxylic acids and alcohols by using aromatic carboxylic acid anhydrides as dehydration condensation agents. In 1994, Prof. Isamu Shiina reported an acidic coupling method using Lewis acid, and, in 2002, a basic esterification using nucleophilic catalyst.