In genetics, shotgun sequencing is a method used for sequencing random DNA strands. It is named by analogy with the rapidly expanding, quasi-random shot grouping of a shotgun.

Genome projects are scientific endeavours that ultimately aim to determine the complete genome sequence of an organism and to annotate protein-coding genes and other important genome-encoded features. The genome sequence of an organism includes the collective DNA sequences of each chromosome in the organism. For a bacterium containing a single chromosome, a genome project will aim to map the sequence of that chromosome. For the human species, whose genome includes 22 pairs of autosomes and 2 sex chromosomes, a complete genome sequence will involve 46 separate chromosome sequences.
A contig is a set of overlapping DNA segments that together represent a consensus region of DNA. In bottom-up sequencing projects, a contig refers to overlapping sequence data (reads); in top-down sequencing projects, contig refers to the overlapping clones that form a physical map of the genome that is used to guide sequencing and assembly. Contigs can thus refer both to overlapping DNA sequences and to overlapping physical segments (fragments) contained in clones depending on the context.
In bioinformatics, sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology might not be able to 'read' whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
In graph theory, an n-dimensional De Bruijn graph of m symbols is a directed graph representing overlaps between sequences of symbols. It has mn vertices, consisting of all possible length-n sequences of the given symbols; the same symbol may appear multiple times in a sequence. For a set of m symbols S = {s1, …, sm}, the set of vertices is:

John Frederick William Birney is joint director of EMBL's European Bioinformatics Institute (EMBL-EBI), in Hinxton, Cambridgeshire and deputy director general of the European Molecular Biology Laboratory (EMBL). He also serves as non-executive director of Genomics England, chair of the Global Alliance for Genomics and Health (GA4GH) and honorary professor of bioinformatics at the University of Cambridge. Birney has made significant contributions to genomics, through his development of innovative bioinformatics and computational biology tools. He previously served as an associate faculty member at the Wellcome Trust Sanger Institute.

In bioinformatics, k-mers are substrings of length contained within a biological sequence. Primarily used within the context of computational genomics and sequence analysis, in which k-mers are composed of nucleotides, k-mers are capitalized upon to assemble DNA sequences, improve heterologous gene expression, identify species in metagenomic samples, and create attenuated vaccines. Usually, the term k-mer refers to all of a sequence's subsequences of length , such that the sequence AGAT would have four monomers, three 2-mers, two 3-mers and one 4-mer (AGAT). More generally, a sequence of length will have k-mers and total possible k-mers, where is number of possible monomers.

RNA-Seq is a sequencing technique which uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment, analyzing the continuously changing cellular transcriptome.
SOAP is a suite of bioinformatics software tools from the BGI Bioinformatics department enabling the assembly, alignment, and analysis of next generation DNA sequencing data. It is particularly suited to short read sequencing data.

In bioinformatics, hybrid genome assembly refers to utilizing various sequencing technologies to achieve the task of assembling a genome from fragmented, sequenced DNA resulting from shotgun sequencing. Genome assembly presents one of the most challenging tasks in genome sequencing as most modern DNA sequencing technologies can only produce reads that are, on average, 25-300 base pairs in length. This is orders of magnitude smaller than the average size of a genome. This assembly is computationally difficult and has some inherent challenges, one of these challenges being that genomes often contain complex tandem repeats of sequences that can be thousands of base pairs in length. These repeats can be long enough that second generation sequencing reads are not long enough to bridge the repeat, and, as such, determining the location of each repeat in the genome can be difficult. Resolving these tandem repeats can be accomplished by utilizing long third generation sequencing reads, such as those obtained using the PacBio RS DNA sequencer. These sequences are, on average, 10,000-15,000 base pairs in length and are long enough to span most repeated regions. Using a hybrid approach to this process can increase the fidelity of assembling tandem repeats by being able to accurately place them along a linear scaffold and make the process more computationally efficient.
De novo transcriptome assembly is the de novo sequence assembly method of creating a transcriptome without the aid of a reference genome.
In DNA sequencing, a read is an inferred sequence of base pairs corresponding to all or part of a single DNA fragment. A typical sequencing experiment involves fragmentation of the genome into millions of molecules, which are size-selected and ligated to adapters. The set of fragments is referred to as a sequencing library, which is sequenced to produce a set of reads.
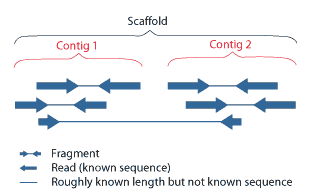
Scaffolding is a technique used in bioinformatics. It is defined as follows:
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.
SPAdes is a genome assembly algorithm which was designed for single cell and multi-cells bacterial data sets. Therefore, it might not be suitable for large genomes projects.
In bioinformatics, a DNA read error occurs when a sequence assembler changes one DNA base for a different base. The reads from the sequence assembler can then be used to create a de Bruijn graph, which can be used in various ways to find errors.
De novo sequence assemblers are a type of program that assembles short nucleotide sequences into longer ones without the use of a reference genome. These are most commonly used in bioinformatic studies to assemble genomes or transcriptomes. Two common types of de novo assemblers are greedy algorithm assemblers and De Bruijn graph assemblers.
A plant genome assembly represents the complete genomic sequence of a plant species, which is assembled into chromosomes and other organelles by using DNA fragments that are obtained from different types of sequencing technology.
Bloom filters are space-efficient probabilistic data structures used to test whether an element is a part of a set. Bloom filters require much less space than other data structures for representing sets, however the downside of Bloom filters is that there is a false positive rate when querying the data structure. Since multiple elements may have the same hash values for a number of hash functions, then there is a probability that querying for a non-existent element may return a positive if another element with the same hash values has been added to the Bloom filter. Assuming that the hash function has equal probability of selecting any index of the Bloom filter, the false positive rate of querying a Bloom filter is a function of the number of bits, number of hash functions and number of elements of the Bloom filter. This allows the user to manage the risk of a getting a false positive by compromising on the space benefits of the Bloom filter.
In bioinformatics, a spaced seed is a pattern of relevant and irrelevant positions in a biosequence and a method of approximate string matching that allows for substitutions. They are a straightforward modification to the earliest heuristic-based alignment efforts that allow for minor differences between the sequences of interest. Spaced seeds have been used in homology search., alignment, assembly, and metagenomics. They are usually represented as a sequence of zeroes and ones, where a one indicates relevance and a zero indicates irrelevance at the given position. Some visual representations use pound signs for relevant and dashes or asterisks for irrelevant positions.




