
A fontanelle is an anatomical feature of the infant human skull comprising soft membranous gaps (sutures) between the cranial bones that make up the calvaria of a fetus or an infant. Fontanelles allow for stretching and deformation of the neurocranium both during birth and later as the brain expands faster than the surrounding bone can grow. Premature complete ossification of the sutures is called craniosynostosis.

Anotia describes a rare congenital deformity that involves the complete absence of the auricle, the outer projected portion of the ear, and narrowing or absence of the ear canal. This contrasts with microtia, in which a small part of the auricle is present. Anotia and microtia may occur unilaterally or bilaterally. This deformity results in conductive hearing loss, deafness.
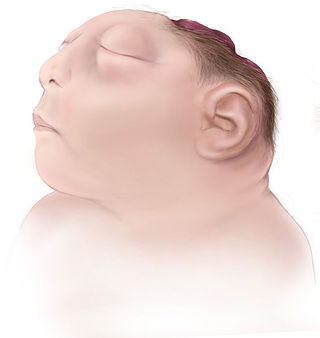
Anencephaly is the absence of a major portion of the brain, skull, and scalp that occurs during embryonic development. It is a cephalic disorder that results from a neural tube defect that occurs when the rostral (head) end of the neural tube fails to close, usually between the 23rd and 26th day following conception. Strictly speaking, the Greek term translates as "without a brain", but it is accepted that children born with this disorder usually only lack a telencephalon, the largest part of the brain consisting mainly of the cerebral hemispheres, including the neocortex, which is responsible for cognition. The remaining structure is usually covered only by a thin layer of membrane—skin, bone, meninges, etc., are all lacking. With very few exceptions, infants with this disorder do not survive longer than a few hours or days after birth.

Colpocephaly is a cephalic disorder involving the disproportionate enlargement of the occipital horns of the lateral ventricles and is usually diagnosed early after birth due to seizures. It is a nonspecific finding and is associated with multiple neurological syndromes, including agenesis of the corpus callosum, Chiari malformation, lissencephaly, and microcephaly. Although the exact cause of colpocephaly is not known yet, it is commonly believed to occur as a result of neuronal migration disorders during early brain development, intrauterine disturbances, perinatal injuries, and other central nervous system disorders. Individuals with colpocephaly have various degrees of motor disabilities, visual defects, spasticity, and moderate to severe intellectual disability. No specific treatment for colpocephaly exists, but patients may undergo certain treatments to improve their motor function or intellectual disability.
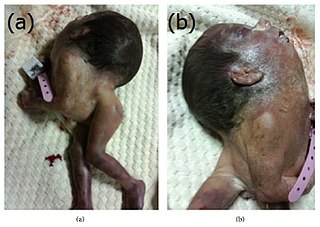
Iniencephaly is a rare type of cephalic disorder characterised by three common characteristics: a defect to the occipital bone, spina bifida of the cervical vertebrae and retroflexion of the head on the cervical spine. Stillbirth is the most common outcome, with a few rare examples of live birth, after which death invariably occurs within a short time.

A birth defect, also known as a congenital disorder, is an abnormal condition that is present at birth regardless of its cause. Birth defects may result in disabilities that may be physical, intellectual, or developmental. The disabilities can range from mild to severe. Birth defects are divided into two main types: structural disorders in which problems are seen with the shape of a body part and functional disorders in which problems exist with how a body part works. Functional disorders include metabolic and degenerative disorders. Some birth defects include both structural and functional disorders.

Omphalocele or omphalocoele also called exomphalos, is a rare abdominal wall defect. Beginning at the 6th week of development, rapid elongation of the gut and increased liver size reduces intra abdominal space, which pushes intestinal loops out of the abdominal cavity. Around 10th week, the intestine returns to the abdominal cavity and the process is completed by the 12th week. Persistence of intestine or the presence of other abdominal viscera in the umbilical cord results in an omphalocele.

Crouzon syndrome is an autosomal dominant genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial arch, which is the precursor of the maxilla and mandible. Because the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects. The syndrome is caused by a mutation in a gene on chromosome 10 that controls the body's production of fibroblast growth factor receptor 2 (FGFR2).

Sirenomelia, also called mermaid syndrome, is a rare congenital deformity in which the legs are fused together, giving the appearance of a mermaid's tail, hence the nickname.
Prenatal development includes the development of the embryo and of the fetus during a viviparous animal's gestation. Prenatal development starts with fertilization, in the germinal stage of embryonic development, and continues in fetal development until birth.

Dandy–Walker malformation (DWM), also known as Dandy–Walker syndrome (DWS), is a rare congenital brain malformation in which the part joining the two hemispheres of the cerebellum does not fully form, and the fourth ventricle and space behind the cerebellum are enlarged with cerebrospinal fluid. Most of those affected develop hydrocephalus within the first year of life, which can present as increasing head size, vomiting, excessive sleepiness, irritability, downward deviation of the eyes and seizures. Other, less common symptoms are generally associated with comorbid genetic conditions and can include congenital heart defects, eye abnormalities, intellectual disability, congenital tumours, other brain defects such as agenesis of the corpus callosum, skeletal abnormalities, an occipital encephalocele or underdeveloped genitalia or kidneys. It is sometimes discovered in adolescents or adults due to mental health problems.

Ablepharon macrostomia syndrome (AMS) is an extremely rare, autosomal dominant genetic disorder characterized by abnormal phenotypic appearances that primarily affect the head and face as well as the skull, skin, fingers and genitals. AMS generally results in abnormal ectoderm-derived structures. The most prominent abnormality is the underdevelopment (microblepharon) or absence of eyelids – signifying the ablepharon aspect of the disease – and a wide, fish-like mouth – macrostomia. Recent scholars and surgeons have called into question the naming of the condition as "Ablepharon" on account of recent investigation and histology showing consistent evidence of at least some eyelid tissue. Infants presenting with AMS may also have malformations of the abdominal wall and nipples. Children with AMS might also experience issues with learning development, language difficulties and intellectual disabilities.
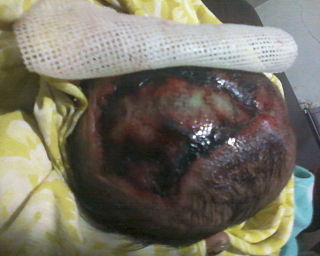
Acrania is a rare congenital disorder that occurs in the human fetus in which the flat bones in the cranial vault are either completely or partially absent. The cerebral hemispheres develop completely but abnormally. The condition is frequently, though not always, associated with anencephaly. The fetus is said to have acrania if it meets the following criteria: the fetus should have a perfectly normal facial bone, a normal cervical column but without the fetal skull and a volume of brain tissue equivalent to at least one-third of the normal brain size.

Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.
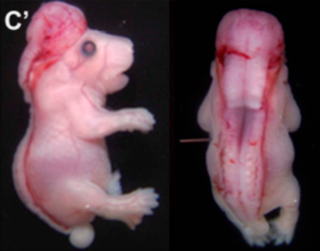
Rachischisis is a developmental birth defect involving the neural tube. This anomaly occurs in utero, when the posterior neuropore of the neural tube fails to close by the 27th intrauterine day. As a consequence the vertebrae overlying the open portion of the spinal cord do not fully form and remain unfused and open, leaving the spinal cord exposed. Patients with rachischisis have motor and sensory deficits, chronic infections, and disturbances in bladder function. This defect often occurs with anencephaly.

Fibrochondrogenesis is a rare autosomal recessive form of osteochondrodysplasia, causing abnormal fibrous development of cartilage and related tissues.
Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.
Limb body wall complex (LBWC) is a rare and severe syndrome of congenital malformations involving craniofacial and abdominal anomalies. LBWC emerges during early fetal development and is fatal. The cause of LBWC is unknown.
XK aprosencephaly is an extremely rare congenital disorder characterized by the absence of the embryonic forebrain. Because the prosencephalon gives way to the cerebral cortex, survival with aprosencephaly is not possible outside utero. The external symptoms are similar to holoprosencephaly, a related disorder, including a smaller than normal head (microcephaly), small eyeballs (microphthalmia), a small mouth (microstomia), anal atresia, and abnormalities of the external genitalia, radius, nostrils, and pharynx (throat).
