The urea cycle (also known as the ornithine cycle) is a cycle of biochemical reactions that produces urea (NH2)2CO from ammonia (NH3). Animals that use this cycle, mainly amphibians and mammals, are called ureotelic.
Canavan disease, or Canavan–Van Bogaert–Bertrand disease, is a rare and fatal autosomal recessive degenerative disease that causes progressive damage to nerve cells and loss of white matter in the brain. It is one of the most common degenerative cerebral diseases of infancy. It is caused by a deficiency of the enzyme aminoacylase 2, and is one of a group of genetic diseases referred to as leukodystrophies. It is characterized by degeneration of myelin in the phospholipid layer insulating the axon of a neuron and is associated with a gene located on human chromosome 17.

Angiotensin-converting enzyme, or ACE, is a central component of the renin–angiotensin system (RAS), which controls blood pressure by regulating the volume of fluids in the body. It converts the hormone angiotensin I to the active vasoconstrictor angiotensin II. Therefore, ACE indirectly increases blood pressure by causing blood vessels to constrict. ACE inhibitors are widely used as pharmaceutical drugs for treatment of cardiovascular diseases.
In molecular biology, biosynthesis is a multi-step, enzyme-catalyzed process where substrates are converted into more complex products in living organisms. In biosynthesis, simple compounds are modified, converted into other compounds, or joined to form macromolecules. This process often consists of metabolic pathways. Some of these biosynthetic pathways are located within a single cellular organelle, while others involve enzymes that are located within multiple cellular organelles. Examples of these biosynthetic pathways include the production of lipid membrane components and nucleotides. Biosynthesis is usually synonymous with anabolism.

Leukodystrophies are a group of, usually, inherited disorders, characterized by degeneration of the white matter in the brain. The word leukodystrophy comes from the Greek roots leuko, "white", dys, "abnormal" and troph, "growth". The leukodystrophies are caused by imperfect growth or development of the glial cells which produce the myelin sheath, the fatty insulating covering around nerve fibers. Leukodystrophies may be classified as hypomyelinating or demyelinating diseases, respectively, depending on whether the damage is present before birth or occurs after. Other demyelinating diseases are usually not congenital and have a toxic or autoimmune cause.
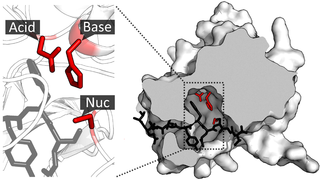
A catalytic triad is a set of three coordinated amino acids that can be found in the active site of some enzymes. Catalytic triads are most commonly found in hydrolase and transferase enzymes. An acid-base-nucleophile triad is a common motif for generating a nucleophilic residue for covalent catalysis. The residues form a charge-relay network to polarise and activate the nucleophile, which attacks the substrate, forming a covalent intermediate which is then hydrolysed to release the product and regenerate free enzyme. The nucleophile is most commonly a serine or cysteine amino acid, but occasionally threonine or even selenocysteine. The 3D structure of the enzyme brings together the triad residues in a precise orientation, even though they may be far apart in the sequence.

β-Glucocerebrosidase is an enzyme with glucosylceramidase activity that cleaves by hydrolysis the β-glycosidic linkage of the chemical glucocerebroside, an intermediate in glycolipid metabolism that is abundant in cell membranes. It is localized in the lysosome, where it remains associated with the lysosomal membrane. β-Glucocerebrosidase is 497 amino acids in length and has a molecular mass of 59,700 Da.
A carboxypeptidase is a protease enzyme that hydrolyzes (cleaves) a peptide bond at the carboxy-terminal (C-terminal) end of a protein or peptide. This is in contrast to an aminopeptidases, which cleave peptide bonds at the N-terminus of proteins. Humans, animals, bacteria and plants contain several types of carboxypeptidases that have diverse functions ranging from catabolism to protein maturation. At least two mechanisms have been discussed.

Hexosaminidase is an enzyme involved in the hydrolysis of terminal N-acetyl-D-hexosamine residues in N-acetyl-β-D-hexosaminides.
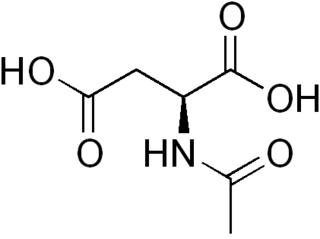
N-Acetylaspartic acid, or N-acetylaspartate (NAA), is a derivative of aspartic acid with a formula of C6H9NO5 and a molecular weight of 175.139.

CAD protein is a trifunctional multi-domain enzyme involved in the first three steps of pyrimidine biosynthesis. De-novo synthesis starts with cytosolic carbamoylphosphate synthetase II which uses glutamine, carbon dioxide and ATP. This enzyme is inhibited by uridine triphosphate.

Myelin protein zero is a single membrane glycoprotein which in humans is encoded by the MPZ gene. P0 is a major structural component of the myelin sheath in the peripheral nervous system (PNS). Myelin protein zero is expressed by Schwann cells and accounts for over 50% of all proteins in the peripheral nervous system, making it the most common protein expressed in the PNS. Mutations in myelin protein zero can cause myelin deficiency and are associated with neuropathies like Charcot–Marie–Tooth disease and Dejerine–Sottas disease.

N-Acetylaspartylglutamic acid is a peptide neurotransmitter and the third-most-prevalent neurotransmitter in the mammalian nervous system. NAAG consists of N-acetylaspartic acid (NAA) and glutamic acid coupled via a peptide bond.
Pantothenate kinase (EC 2.7.1.33, PanK; CoaA) is the first enzyme in the Coenzyme A (CoA) biosynthetic pathway. It phosphorylates pantothenate (vitamin B5) to form 4'-phosphopantothenate at the expense of a molecule of adenosine triphosphate (ATP). It is the rate-limiting step in the biosynthesis of CoA.
Carboxypeptidase A usually refers to the pancreatic exopeptidase that hydrolyzes peptide bonds of C-terminal residues with aromatic or aliphatic side-chains. Most scientists in the field now refer to this enzyme as CPA1, and to a related pancreatic carboxypeptidase as CPA2.

Palmitoyl protein hydrolase/thioesterases is an enzyme (EC 3.1.2.22) that removes thioester-linked fatty acyl groups such as palmitate from modified cysteine residues in proteins or peptides during lysosomal degradation. It catalyzes the reaction
In enzymology, an aminoacylase (EC 3.5.1.14) is an enzyme that catalyzes the chemical reaction

Protein O-GlcNAcase (EC 3.2.1.169, OGA, glycoside hydrolase O-GlcNAcase, O-GlcNAcase, BtGH84, O-GlcNAc hydrolase) is an enzyme with systematic name (protein)-3-O-(N-acetyl-D-glucosaminyl)-L-serine/threonine N-acetylglucosaminyl hydrolase. OGA is encoded by the OGA gene. This enzyme catalyses the removal of the O-GlcNAc post-translational modification in the following chemical reaction:
- [protein]-3-O-(N-acetyl-β-D-glucosaminyl)-L-serine + H2O ⇌ [protein]-L-serine + N-acetyl-D-glucosamine
- [protein]-3-O-(N-acetyl-β-D-glucosaminyl)-L-threonine + H2O ⇌ [protein]-L-threonine + N-acetyl-D-glucosamine

alpha/beta-Hydrolase domain containing 12 (ABHD12) is a serine hydrolase encoded by the ABHD12 gene that participates in the breakdown of the endocannabinoid neurotransmitter 2-arachidonylglycerol (2-AG) in the central nervous system. It is responsible for about 9% of brain 2-AG hydrolysis. Together, ABHD12 along with two other enzymes, monoacylglycerol lipase (MAGL) and ABHD6, control 99% of 2-AG hydrolysis in the brain. ABHD12 also serves as a lysophospholipase and metabolizes lysophosphatidylserine (LPS).

Spongy degeneration of the central nervous system, also known as Canavan's disease, Van Bogaert-Bertrand type or Aspartoacylase (AspA) deficiency, is a rare autosomal recessive neurodegenerative disorder. It belongs to a group of genetic disorders known as leukodystrophies, where the growth and maintenance of myelin sheath in the central nervous system (CNS) are impaired. There are three types of spongy degeneration: infantile, congenital and juvenile, with juvenile being the most severe type. Common symptoms in infants include lack of motor skills, weak muscle tone, and macrocephaly. It may also be accompanied by difficulties in feeding and swallowing, seizures and sleep disturbances. Affected children typically die before the age of 10, but life expectancy can vary.








