
Fluorescence is one of two kinds of emission of light by a substance that has absorbed light or other electromagnetic radiation. Fluorescence involves no change in electron spin multiplicity and generally it immediately follows absorption; phosphorescence involves spin change and is delayed. Thus fluorescent materials generally cease to glow nearly immediately when the radiation source stops, while phosphorescent materials, which continue to emit light for some time after.

Microscopy is the technical field of using microscopes to view objects and areas of objects that cannot be seen with the naked eye. There are three well-known branches of microscopy: optical, electron, and scanning probe microscopy, along with the emerging field of X-ray microscopy.

Immunofluorescence(IF) is a light microscopy-based technique that allows detection and localization of a wide variety of target biomolecules within a cell or tissue at a quantitative level. The technique utilizes the binding specificity of antibodies and antigens. The specific region an antibody recognizes on an antigen is called an epitope. Several antibodies can recognize the same epitope but differ in their binding affinity. The antibody with the higher affinity for a specific epitope will surpass antibodies with a lower affinity for the same epitope.
A total internal reflection fluorescence microscope (TIRFM) is a type of microscope with which a thin region of a specimen, usually less than 200 nanometers can be observed.

A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.

Confocal microscopy, most frequently confocal laser scanning microscopy (CLSM) or laser scanning confocal microscopy (LSCM), is an optical imaging technique for increasing optical resolution and contrast of a micrograph by means of using a spatial pinhole to block out-of-focus light in image formation. Capturing multiple two-dimensional images at different depths in a sample enables the reconstruction of three-dimensional structures within an object. This technique is used extensively in the scientific and industrial communities and typical applications are in life sciences, semiconductor inspection and materials science.

Fluorescence in situ hybridization (FISH) is a molecular cytogenetic technique that uses fluorescent probes that bind to only particular parts of a nucleic acid sequence with a high degree of sequence complementarity. It was developed by biomedical researchers in the early 1980s to detect and localize the presence or absence of specific DNA sequences on chromosomes. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes. FISH is often used for finding specific features in DNA for use in genetic counseling, medicine, and species identification. FISH can also be used to detect and localize specific RNA targets in cells, circulating tumor cells, and tissue samples. In this context, it can help define the spatial-temporal patterns of gene expression within cells and tissues.

Two-photon excitation microscopy is a fluorescence imaging technique that is particularly well-suited to image scattering living tissue of up to about one millimeter in thickness. Unlike traditional fluorescence microscopy, where the excitation wavelength is shorter than the emission wavelength, two-photon excitation requires simultaneous excitation by two photons with longer wavelength than the emitted light. The laser is focused onto a specific location in the tissue and scanned across the sample to sequentially produce the image. Due to the non-linearity of two-photon excitation, mainly fluorophores in the micrometer-sized focus of the laser beam are excited, which results in the spatial resolution of the image. This contrasts with confocal microscopy, where the spatial resolution is produced by the interaction of excitation focus and the confined detection with a pinhole.
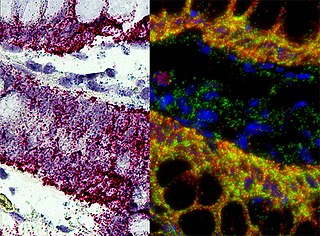
In situ hybridization (ISH) is a type of hybridization that uses a labeled complementary DNA, RNA or modified nucleic acids strand to localize a specific DNA or RNA sequence in a portion or section of tissue or if the tissue is small enough, in the entire tissue, in cells, and in circulating tumor cells (CTCs). This is distinct from immunohistochemistry, which usually localizes proteins in tissue sections.
Fluorescence correlation spectroscopy (FCS) is a statistical analysis, via time correlation, of stationary fluctuations of the fluorescence intensity. Its theoretical underpinning originated from L. Onsager's regression hypothesis. The analysis provides kinetic parameters of the physical processes underlying the fluctuations. One of the interesting applications of this is an analysis of the concentration fluctuations of fluorescent particles (molecules) in solution. In this application, the fluorescence emitted from a very tiny space in solution containing a small number of fluorescent particles (molecules) is observed. The fluorescence intensity is fluctuating due to Brownian motion of the particles. In other words, the number of the particles in the sub-space defined by the optical system is randomly changing around the average number. The analysis gives the average number of fluorescent particles and average diffusion time, when the particle is passing through the space. Eventually, both the concentration and size of the particle (molecule) are determined. Both parameters are important in biochemical research, biophysics, and chemistry.

In optics, photobleaching is the photochemical alteration of a dye or a fluorophore molecule such that it is permanently unable to fluoresce. This is caused by cleaving of covalent bonds or non-specific reactions between the fluorophore and surrounding molecules. Such irreversible modifications in covalent bonds are caused by transition from a singlet state to the triplet state of the fluorophores. The number of excitation cycles to achieve full bleaching varies. In microscopy, photobleaching may complicate the observation of fluorescent molecules, since they will eventually be destroyed by the light exposure necessary to stimulate them into fluorescing. This is especially problematic in time-lapse microscopy.

Second-harmonic imaging microscopy (SHIM) is based on a nonlinear optical effect known as second-harmonic generation (SHG). SHIM has been established as a viable microscope imaging contrast mechanism for visualization of cell and tissue structure and function. A second-harmonic microscope obtains contrasts from variations in a specimen's ability to generate second-harmonic light from the incident light while a conventional optical microscope obtains its contrast by detecting variations in optical density, path length, or refractive index of the specimen. SHG requires intense laser light passing through a material with a noncentrosymmetric molecular structure, either inherent or induced externally, for example by an electric field.

Vertico spatially modulated illumination (Vertico-SMI) is the fastest light microscope for the 3D analysis of complete cells in the nanometer range. It is based on two technologies developed in 1996, SMI and SPDM. The effective optical resolution of this optical nanoscope has reached the vicinity of 5 nm in 2D and 40 nm in 3D, greatly surpassing the λ/2 resolution limit applying to standard microscopy using transmission or reflection of natural light according to the Abbe resolution limit That limit had been determined by Ernst Abbe in 1873 and governs the achievable resolution limit of microscopes using conventional techniques.

Fluorescence is used in the life sciences generally as a non-destructive way of tracking or analysing biological molecules. Some proteins or small molecules in cells are naturally fluorescent, which is called intrinsic fluorescence or autofluorescence. Alternatively, specific or general proteins, nucleic acids, lipids or small molecules can be "labelled" with an extrinsic fluorophore, a fluorescent dye which can be a small molecule, protein or quantum dot. Several techniques exist to exploit additional properties of fluorophores, such as fluorescence resonance energy transfer, where the energy is passed non-radiatively to a particular neighbouring dye, allowing proximity or protein activation to be detected; another is the change in properties, such as intensity, of certain dyes depending on their environment allowing their use in structural studies.
Super-resolution microscopy is a series of techniques in optical microscopy that allow such images to have resolutions higher than those imposed by the diffraction limit, which is due to the diffraction of light. Super-resolution imaging techniques rely on the near-field or on the far-field. Among techniques that rely on the latter are those that improve the resolution only modestly beyond the diffraction-limit, such as confocal microscopy with closed pinhole or aided by computational methods such as deconvolution or detector-based pixel reassignment, the 4Pi microscope, and structured-illumination microscopy technologies such as SIM and SMI.
Photo-activated localization microscopy and stochastic optical reconstruction microscopy (STORM) are widefield fluorescence microscopy imaging methods that allow obtaining images with a resolution beyond the diffraction limit. The methods were proposed in 2006 in the wake of a general emergence of optical super-resolution microscopy methods, and were featured as Methods of the Year for 2008 by the Nature Methods journal. The development of PALM as a targeted biophysical imaging method was largely prompted by the discovery of new species and the engineering of mutants of fluorescent proteins displaying a controllable photochromism, such as photo-activatible GFP. However, the concomitant development of STORM, sharing the same fundamental principle, originally made use of paired cyanine dyes. One molecule of the pair, when excited near its absorption maximum, serves to reactivate the other molecule to the fluorescent state.

Light sheet fluorescence microscopy (LSFM) is a fluorescence microscopy technique with an intermediate-to-high optical resolution, but good optical sectioning capabilities and high speed. In contrast to epifluorescence microscopy only a thin slice of the sample is illuminated perpendicularly to the direction of observation. For illumination, a laser light-sheet is used, i.e. a laser beam which is focused only in one direction. A second method uses a circular beam scanned in one direction to create the lightsheet. As only the actually observed section is illuminated, this method reduces the photodamage and stress induced on a living sample. Also the good optical sectioning capability reduces the background signal and thus creates images with higher contrast, comparable to confocal microscopy. Because light sheet fluorescence microscopy scans samples by using a plane of light instead of a point, it can acquire images at speeds 100 to 1,000 times faster than those offered by point-scanning methods.
Super-resolution dipole orientation mapping (SDOM) is a form of fluorescence polarization microscopy (FPM) that achieved super resolution through polarization demodulation. It was first described by Karl Zhanghao and others in 2016. Fluorescence polarization (FP) is related to the dipole orientation of chromophores, making fluorescence polarization microscopy possible to reveal structures and functions of tagged cellular organelles and biological macromolecules. In addition to fluorescence intensity, wavelength, and lifetime, the fourth dimension of fluorescence—polarization—can also provide intensity modulation without the restriction to specific fluorophores; its investigation in super-resolution microscopy is still in its infancy.
Microscopy with UV Surface Excitation (MUSE) is a novel microscopy method that utilizes the shallow penetration of UV photons excitation. Compared to conventional microscopes, which usually require sectioning to exclude blurred signals from outside of the focal plane, MUSE's low penetration depth limits the excitation volume to a thin layer, and removes the tissue sectioning requirement. The entire signal collected is the desired light, and all photons collected contribute to the image formation.
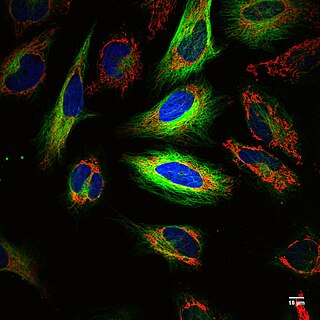
Fluorescence imaging is a type of non-invasive imaging technique that can help visualize biological processes taking place in a living organism. Images can be produced from a variety of methods including: microscopy, imaging probes, and spectroscopy.



