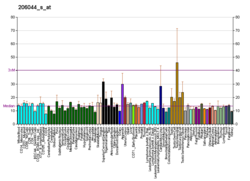
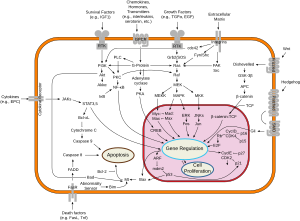
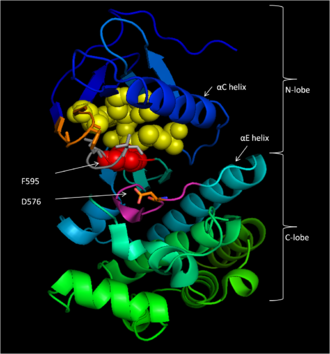
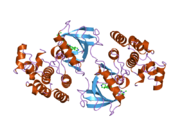A protein kinase is a kinase which selectively modifies other proteins by covalently adding phosphates to them (phosphorylation) as opposed to kinases which modify lipids, carbohydrates, or other molecules. Phosphorylation usually results in a functional change of the target protein (substrate) by changing enzyme activity, cellular location, or association with other proteins. The human genome contains about 500 protein kinase genes and they constitute about 2% of all human genes. There are two main types of protein kinase. The great majority are serine/threonine kinases, which phosphorylate the hydroxyl groups of serines and threonines in their targets. Most of the others are tyrosine kinases, although additional types exist. Protein kinases are also found in bacteria and plants. Up to 30% of all human proteins may be modified by kinase activity, and kinases are known to regulate the majority of cellular pathways, especially those involved in signal transduction.

In biochemistry, a kinase is an enzyme that catalyzes the transfer of phosphate groups from high-energy, phosphate-donating molecules to specific substrates. This process is known as phosphorylation, where the high-energy ATP molecule donates a phosphate group to the substrate molecule. As a result, kinase produces a phosphorylated substrate and ADP. Conversely, it is referred to as dephosphorylation when the phosphorylated substrate donates a phosphate group and ADP gains a phosphate group. These two processes, phosphorylation and dephosphorylation, occur four times during glycolysis.
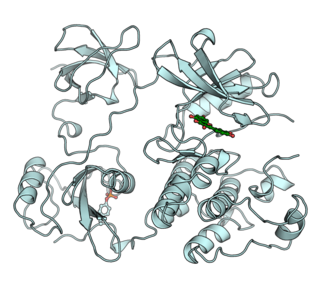
A tyrosine kinase is an enzyme that can transfer a phosphate group from ATP to the tyrosine residues of specific proteins inside a cell. It functions as an "on" or "off" switch in many cellular functions.

Cyclin-dependent kinases (CDKs) are a predominant group of serine/threonine protein kinases involved in the regulation of the cell cycle and its progression, ensuring the integrity and functionality of cellular machinery. These regulatory enzymes play a crucial role in the regulation of eukaryotic cell cycle and transcription, as well as DNA repair, metabolism, and epigenetic regulation, in response to several extracellular and intracellular signals. They are present in all known eukaryotes, and their regulatory function in the cell cycle has been evolutionarily conserved. The catalytic activities of CDKs are regulated by interactions with CDK inhibitors (CKIs) and regulatory subunits known as cyclins. Cyclins have no enzymatic activity themselves, but they become active once they bind to CDKs. Without cyclin, CDK is less active than in the cyclin-CDK heterodimer complex. CDKs phosphorylate proteins on serine (S) or threonine (T) residues. The specificity of CDKs for their substrates is defined by the S/T-P-X-K/R sequence, where S/T is the phosphorylation site, P is proline, X is any amino acid, and the sequence ends with lysine (K) or arginine (R). This motif ensures CDKs accurately target and modify proteins, crucial for regulating cell cycle and other functions. Deregulation of the CDK activity is linked to various pathologies, including cancer, neurodegenerative diseases, and stroke.

RAF proto-oncogene serine/threonine-protein kinase, also known as proto-oncogene c-RAF or simply c-Raf or even Raf-1, is an enzyme that in humans is encoded by the RAF1 gene. The c-Raf protein is part of the ERK1/2 pathway as a MAP kinase (MAP3K) that functions downstream of the Ras subfamily of membrane associated GTPases. C-Raf is a member of the Raf kinase family of serine/threonine-specific protein kinases, from the TKL (Tyrosine-kinase-like) group of kinases.

A serine/threonine protein kinase is a kinase enzyme, in particular a protein kinase, that phosphorylates the OH group of the amino-acid residues serine or threonine, which have similar side chains. At least 350 of the 500+ human protein kinases are serine/threonine kinases (STK).

A cyclin-dependent kinase inhibitor protein(also known as CKIs, CDIs, or CDKIs) is a protein which inhibits the enzyme cyclin-dependent kinase (CDK) and Cyclin activity by stopping the cell cycle if there are unfavorable conditions, therefore, acting as tumor suppressors. Cell cycle progression is stopped by Cyclin-dependent kinase inhibitor protein at the G1 phase. CKIs are vital proteins within the control system that point out whether the process of DNA synthesis, mitosis, and cytokines control one another. If a malfunction prevents the successful completion of DNA synthesis during the G1 phase, a signal is sent to delay or stop the progression to the S phase. Cyclin-dependent kinase inhibitor proteins are essential in the regulation of the cell cycle. If cell mutations surpass the cell cycle checkpoints during cell cycle regulation, it can result in various types of cancer.
The ErbB family of proteins contains four receptor tyrosine kinases, structurally related to the epidermal growth factor receptor (EGFR), its first discovered member. In humans, the family includes Her1, Her2 (ErbB2), Her3 (ErbB3), and Her4 (ErbB4). The gene symbol, ErbB, is derived from the name of a viral oncogene to which these receptors are homologous: erythroblastic leukemia viral oncogene. Insufficient ErbB signaling in humans is associated with the development of neurodegenerative diseases, such as multiple sclerosis and Alzheimer's disease, while excessive ErbB signaling is associated with the development of a wide variety of types of solid tumor.
The IκB kinase is an enzyme complex that is involved in propagating the cellular response to inflammation, specifically the regulation of lymphocytes.
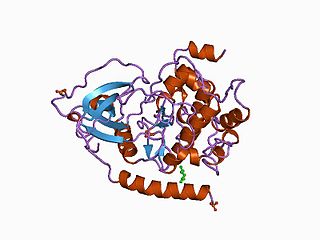
The protein kinase domain is a structurally conserved protein domain containing the catalytic function of protein kinases. Protein kinases are a group of enzymes that move a phosphate group onto proteins, in a process called phosphorylation. This functions as an on/off switch for many cellular processes, including metabolism, transcription, cell cycle progression, cytoskeletal rearrangement and cell movement, apoptosis, and differentiation. They also function in embryonic development, physiological responses, and in the nervous and immune system. Abnormal phosphorylation causes many human diseases, including cancer, and drugs that affect phosphorylation can treat those diseases.
A non-receptor tyrosine kinase (nRTK) is a cytosolic enzyme that is responsible for catalysing the transfer of a phosphate group from a nucleoside triphosphate donor, such as ATP, to tyrosine residues in proteins. Non-receptor tyrosine kinases are a subgroup of protein family tyrosine kinases, enzymes that can transfer the phosphate group from ATP to a tyrosine residue of a protein (phosphorylation). These enzymes regulate many cellular functions by switching on or switching off other enzymes in a cell.
The Walker A and Walker B motifs are protein sequence motifs, known to have highly conserved three-dimensional structures. These were first reported in ATP-binding proteins by Walker and co-workers in 1982.

Vemurafenib (INN), sold under the brand name Zelboraf, is a medication used for the treatment of late-stage melanoma. It is an inhibitor of the B-Raf enzyme and was developed by Plexxikon.
Bcr-Abl tyrosine-kinase inhibitors (TKI) are the first-line therapy for most patients with chronic myelogenous leukemia (CML). More than 90% of CML cases are caused by a chromosomal abnormality that results in the formation of a so-called Philadelphia chromosome. This abnormality was discovered by Peter Nowell in 1960 and is a consequence of fusion between the Abelson (Abl) tyrosine kinase gene at chromosome 9 and the break point cluster (Bcr) gene at chromosome 22, resulting in a chimeric oncogene (Bcr-Abl) and a constitutively active Bcr-Abl tyrosine kinase that has been implicated in the pathogenesis of CML. Compounds have been developed to selectively inhibit the tyrosine kinase.

Balanol is a fungal metabolite produced by the fungus Verticillium balanoides. It is a potent inhibitor of the serine/threonine kinases protein kinase A (PKA) and protein kinase C (PKC), binding in a similar manner with that of ATP. Balanol was discovered in 1993 in the search for novel inhibitors of PKC, a member of a family of serine/threonine kinases whose overactivation is associated with numerous human diseases of signal transduction including cancer. However, much of the research on balanol focuses on how chemical modifications of the molecular structure affect binding to PKA. Indeed, balanol, its chemically altered analogs, and their interactions with PKA in particular are used to illuminate the roles of selectivity and protein flexibility in the inhibition of kinases. For instance, the X-ray crystal structure of balanol in complex with PKA was used in order to confer selectivity and to improve pharmacological efficacy of inhibitors of the H. sapiens Akt (PKB), another serine/threonine protein kinase implicated in the proper functioning of many cellular processes.

Autophosphorylation is a type of post-translational modification of proteins. It is generally defined as the phosphorylation of the kinase by itself. In eukaryotes, this process occurs by the addition of a phosphate group to serine, threonine or tyrosine residues within protein kinases, normally to regulate the catalytic activity. Autophosphorylation may occur when a kinases' own active site catalyzes the phosphorylation reaction, or when another kinase of the same type provides the active site that carries out the chemistry. The latter often occurs when kinase molecules dimerize. In general, the phosphate groups introduced are gamma phosphates from nucleoside triphosphates, most commonly ATP.
A MEK inhibitor is a chemical or drug that inhibits the mitogen-activated protein kinase kinase enzymes MEK1 and/or MEK2. They can be used to affect the MAPK/ERK pathway which is often overactive in some cancers.

Encorafenib, sold under the brand name Braftovi, is a medication for the treatment of certain melanoma cancers. It is a small molecule BRAF inhibitor that targets key enzymes in the MAPK signaling pathway. This pathway occurs in many different cancers including melanoma and colorectal cancers. The substance was being developed by Novartis and then by Array BioPharma. In June 2018, it was approved by the FDA in combination with binimetinib for the treatment of patients with unresectable or metastatic BRAF V600E or V600K mutation-positive melanoma.

Binimetinib, sold under the brand name Mektovi, is an anti-cancer medication used to treat various cancers. Binimetinib is a selective inhibitor of MEK, a central kinase in the tumor-promoting MAPK pathway. Inappropriate activation of the pathway has been shown to occur in many cancers. In June 2018 it was approved by the FDA in combination with encorafenib for the treatment of patients with unresectable or metastatic BRAF V600E or V600K mutation-positive melanoma. In October 2023, it was approved by the FDA for treatment of NSCLC with a BRAF V600E mutation in combination with encorafenib. It was developed by Array Biopharma.
V600E is a mutation of the BRAF gene in which valine (V) is substituted by glutamic acid (E) at amino acid 600. It is a driver mutation in a proportion of certain diagnoses, including melanoma, hairy cell leukemia, papillary thyroid carcinoma, colorectal cancer, non-small-cell lung cancer, Langerhans cell histiocytosis, Erdheim–Chester disease and ameloblastoma.