
Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.

Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.

Noonan syndrome (NS) is a genetic disorder that may present with mildly unusual facial features, short height, congenital heart disease, bleeding problems, and skeletal malformations. Facial features include widely spaced eyes, light-colored eyes, low-set ears, a short neck, and a small lower jaw. Heart problems may include pulmonary valve stenosis. The breast bone may either protrude or be sunken, while the spine may be abnormally curved. Intelligence is often normal. Complications of NS can include leukemia.

Coffin–Lowry syndrome is a genetic disorder that is X-linked dominant and which causes severe mental problems sometimes associated with abnormalities of growth, cardiac abnormalities, kyphoscoliosis, as well as auditory and visual abnormalities.

Costello syndrome, also called faciocutaneoskeletal syndrome or FCS syndrome, is a rare genetic disorder that affects many parts of the body. It is characterized by delayed development and intellectual disabilities, distinctive facial features, unusually flexible joints, and loose folds of extra skin, especially on the hands and feet. Heart abnormalities are common, including a very fast heartbeat (tachycardia), structural heart defects, and overgrowth of the heart muscle. Infants with Costello syndrome may be large at birth, but grow more slowly than other children and have difficulty feeding. Later in life, people with this condition have relatively short stature and many have reduced levels of growth hormones. It is a RASopathy.
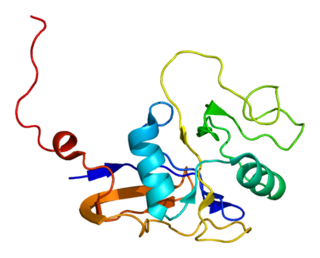
GTPase HRas, from "Harvey Rat sarcoma virus", also known as transforming protein p21 is an enzyme that in humans is encoded by the HRAS gene. The HRAS gene is located on the short (p) arm of chromosome 11 at position 15.5, from base pair 522,241 to base pair 525,549. HRas is a small G protein in the Ras subfamily of the Ras superfamily of small GTPases. Once bound to Guanosine triphosphate, H-Ras will activate a Raf kinase like c-Raf, the next step in the MAPK/ERK pathway.
The MAPK/ERK pathway is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell.

RAF proto-oncogene serine/threonine-protein kinase, also known as proto-oncogene c-RAF or simply c-Raf or even Raf-1, is an enzyme that in humans is encoded by the RAF1 gene. The c-Raf protein is part of the ERK1/2 pathway as a MAP kinase (MAP3K) that functions downstream of the Ras subfamily of membrane associated GTPases. C-Raf is a member of the Raf kinase family of serine/threonine-specific protein kinases, from the TKL (Tyrosine-kinase-like) group of kinases.

Tyrosine-protein phosphatase non-receptor type 11 (PTPN11) also known as protein-tyrosine phosphatase 1D (PTP-1D), Src homology region 2 domain-containing phosphatase-2 (SHP-2), or protein-tyrosine phosphatase 2C (PTP-2C) is an enzyme that in humans is encoded by the PTPN11 gene. PTPN11 is a protein tyrosine phosphatase (PTP) Shp2.

Noonan syndrome with multiple lentigines (NSML) which is part of a group called Ras/MAPK pathway syndromes, is a rare autosomal dominant, multisystem disease caused by a mutation in the protein tyrosine phosphatase, non-receptor type 11 gene (PTPN11). The disease is a complex of features, mostly involving the skin, skeletal and cardiovascular systems, which may or may not be present in all patients. The nature of how the mutation causes each of the condition's symptoms is not well known; however, research is ongoing. It is a RASopathy.
In cell signalling, Son of Sevenless (SOS) refers to a set of genes encoding guanine nucleotide exchange factors that act on the Ras subfamily of small GTPases.
Aarskog–Scott syndrome (AAS) is a rare disease inherited as X-linked and characterized by short stature, facial abnormalities, skeletal and genital anomalies. This condition mainly affects males, although females may have mild features of the syndrome.

In molecular biology, ribosomal s6 kinase (rsk) is a family of protein kinases involved in signal transduction. There are two subfamilies of rsk, p90rsk, also known as MAPK-activated protein kinase-1 (MAPKAP-K1), and p70rsk, also known as S6-H1 Kinase or simply S6 Kinase. There are three variants of p90rsk in humans, rsk 1-3. Rsks are serine/threonine kinases and are activated by the MAPK/ERK pathway. There are two known mammalian homologues of S6 Kinase: S6K1 and S6K2.
Ablepharon macrostomia syndrome (AMS) is an extremely rare, autosomal dominant genetic disorder characterized by abnormal phenotypic appearances that primarily affect the head and face as well as the skull, skin, fingers and genitals. AMS generally results in abnormal ectoderm-derived structures. The most prominent abnormality is the underdevelopment (microblepharon) or absence of eyelids – signifying the ablepharon aspect of the disease – and a wide, fish-like mouth – macrostomia. Infants presenting with AMS may also have malformations of the abdominal wall and nipples. Children with AMS might also experience issues with learning development, language difficulties and intellectual disabilities.

protein S6 kinase, 90kDa, polypeptide 3, also s RPS6KA3, is an enzyme that in humans is encoded by the RPS6KA3 gene.
Neuro-cardio-facial-cutaneous-syndromes (NCFC), is a group of developmental disorders with a genetic ground, affecting the nervous system, circulatory system, (cranio)facial and cutaneous development. These four parts are the common denominator for the syndromes, but are mostly accompanied by disturbances in other parts of the body.
Parkes Weber syndrome (PWS) is a congenital disorder of the vascular system. It is an extremely rare condition, and its exact prevalence is unknown. It is named after British dermatologist Frederick Parkes Weber, who first described the syndrome in 1907.
The RASopathies are developmental syndromes caused by germline mutations in genes that alter the Ras subfamily and mitogen-activated protein kinases that control signal transduction, including:
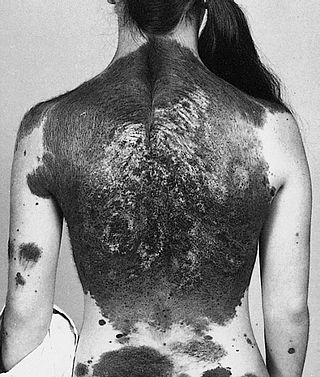
Neurocutaneous melanosis is a congenital disorder characterized by the presence of congenital melanocytic nevi on the skin and melanocytic tumors in the leptomeninges of the central nervous system. These lesions may occur in the amygdala, cerebellum, cerebrum, pons and spinal cord of patients. Although typically asymptomatic, malignancy occurs in the form of leptomeningeal melanoma in over half of patients. Regardless of the presence of malignancy, patients with symptomatic neurocutaneous melanosis generally have a poor prognosis with few treatment options. The pathogenesis of neurocutaneous melanosis is believed to be related to the abnormal postzygotic development of melanoblasts and mutations of the NRAS gene.
Acromesomelic dysplasia is a rare skeletal disorder that causes abnormal bone and cartilage development, leading to shortening of the forearms, lower legs, hands, feet, fingers, and toes. Five different genetic mutations have been implicated in the disorder. Treatment is individualized but is generally aimed at palliating symptoms, for example, treatment of kyphosis and lumbar hyperlordosis.
