A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

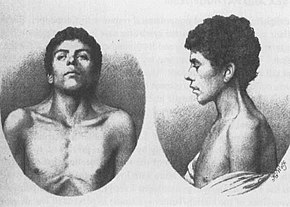
Waardenburg syndrome is a group of rare genetic conditions characterised by at least some degree of congenital hearing loss and pigmentation deficiencies, which can include bright blue eyes, a white forelock or patches of light skin. These basic features constitute type 2 of the condition; in type 1, there is also a wider gap between the inner corners of the eyes called telecanthus, or dystopia canthorum. In type 3, which is rare, the arms and hands are also malformed, with permanent finger contractures or fused fingers, while in type 4, the person also has Hirschsprung's disease. There also exist at least two types that can result in central nervous system (CNS) symptoms such as developmental delay and muscle tone abnormalities.

Kabuki syndrome is a rare congenital disorder of genetic origin. It affects multiple parts of the body, with varying symptoms and severity, although the most common is the characteristic facial appearance.

Alagille syndrome (ALGS) is a genetic disorder that affects primarily the liver and the heart. Problems associated with the disorder generally become evident in infancy or early childhood. The disorder is inherited in an autosomal dominant pattern, and the estimated prevalence of Alagille syndrome is 1 in every 30,000 to 1 in every 40,000 live births. It is named after the French pediatrician Daniel Alagille, who first described the condition in 1969. Children with Alagille syndrome live to the age of 18 in about 90% of the cases.

CHARGE syndrome is a rare syndrome caused by a genetic disorder. First described in 1979, the acronym "CHARGE" came into use for newborn children with the congenital features of coloboma of the eye, heart defects, atresia of the nasal choanae, restricted growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness. These features are no longer used in making a diagnosis of CHARGE syndrome, but the name remains. About two thirds of cases are due to a CHD7 mutation. CHARGE syndrome occurs only in 0.1–1.2 per 10,000 live births; as of 2009, it was the leading cause of congenital deafblindness in the US.

Fraser syndrome is an autosomal recessive congenital disorder, identified by several developmental anomalies. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.
Barakat syndrome is a rare disease characterized by hypoparathyroidism, sensorineural deafness and renal disease, and hence also known as HDR syndrome. It is an autosomal dominant condition with incomplete penetrance and variable expressivity that was first described by Amin J. Barakat et al. in 1977.

Simpson–Golabi–Behmel syndrome (SGBS) is a rare inherited congenital disorder that can cause craniofacial, skeletal, vascular, cardiac, and renal abnormalities. There is a high prevalence of cancer associated in those with SGBS which includes wilms tumors, neuroblastoma, tumors of the adrenal gland, liver, lungs and abdominal organs. The syndrome is inherited in an X-linked recessive manner. Females that possess one copy of the mutation are considered to be carriers of the syndrome but may still express varying degrees of the phenotype, suffering mild to severe malady. Males experience a higher likelihood of fetal death.

MERRF syndrome is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.

Noonan syndrome with multiple lentigines (NSML) which is part of a group called Ras/MAPK pathway syndromes, is a rare autosomal dominant, multisystem disease caused by a mutation in the protein tyrosine phosphatase, non-receptor type 11 gene (PTPN11). The disease is a complex of features, mostly involving the skin, skeletal and cardiovascular systems, which may or may not be present in all patients. The nature of how the mutation causes each of the condition's symptoms is not well known; however, research is ongoing. It is a RASopathy.
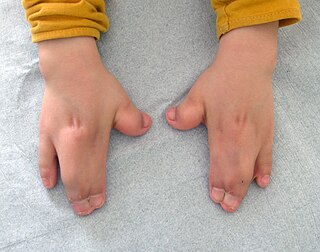
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly). Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth. Syndactyly occurs when digits of the hands or feet are fused together. When polydactyly is also present, the classification is acrocephalopolysyndactyly. Polydactyly occurs when the hands or feet possess additional digits. Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.

Axenfeld–Rieger syndrome is a rare autosomal dominant disorder, which affects the development of the teeth, eyes, and abdominal region.

3C syndrome is a rare condition whose symptoms include heart defects, cerebellar hypoplasia, and cranial dysmorphism. It was first described in the medical literature in 1987 by Ritscher and Schinzel, for whom the disorder is sometimes named.

Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.
The RASopathies are a group of developmental syndromes caused by germline mutations in genes belonging to the Ras/MAPK pathway. Common features include intellectual disability, congenital heart defects, skin abnormalities, and craniofacial abnormalities.
Floating–Harbor syndrome, also known as Pelletier–Leisti syndrome, is a rare disease with fewer than 50 cases described in the literature. It is usually diagnosed in early childhood and is characterized by the triad of proportionate short stature with delayed bone age, characteristic facial appearance, and delayed speech development. Although its cause is unknown, it is thought to result from genetic mutation, and diagnosis is established by the presence of a heterozygous SRCAP mutation in those with clinical findings of FHS.
XK aprosencephaly is an extremely rare congenital disorder characterized by the absence of the embryonic forebrain. Because the prosencephalon gives way to the cerebral cortex, survival with aprosencephaly is not possible outside utero. The external symptoms are similar to holoprosencephaly, a related disorder, including a smaller than normal head (microcephaly), small eyeballs (microphthalmia), a small mouth (microstomia), anal atresia, and abnormalities of the external genitalia, radius, nostrils, and pharynx (throat).

Waardenburg syndrome type 1 is a congenital disorder that caused by a mutation in the PAX3 gene that results in abnormal development in the neural crest during early development. Type 1 results in early graying and white forelock and a notable distance between the eyes, noted as dystopia canthorum. Common symptoms of the disease also includes non-progressive hearing loss in majority of patients with type 1. Patients can display complete or partial heterochromia and hypoplastic blue irides and congenital leukemia.

Pentasomy X, also known as 49,XXXXX, is a chromosomal disorder in which a female has five, rather than two, copies of the X chromosome. Pentasomy X is associated with short stature, intellectual disability, characteristic facial features, heart defects, skeletal anomalies, and pubertal and reproductive abnormalities. The condition is exceptionally rare, with an estimated prevalence between 1 in 85,000 and 1 in 250,000.
Filippi syndrome, also known as Syndactyly Type I with Microcephaly and Mental Retardation, is a very rare autosomal recessive genetic disease. Only a very limited number of cases have been reported to date. Filippi Syndrome is associated with diverse symptoms of varying severity across affected individuals, for example malformation of digits, craniofacial abnormalities, intellectual disability, and growth retardation. The diagnosis of Filippi Syndrome can be done through clinical observation, radiography, and genetic testing. Filippi Syndrome cannot be cured directly as of 2022, hence the main focus of treatments is on tackling the symptoms observed on affected individuals. It was first reported in 1985.