Insulin resistance (IR) is a pathological condition in which cells either fail to respond normally to the hormone insulin or downregulate insulin receptors in response to hyperinsulinemia.
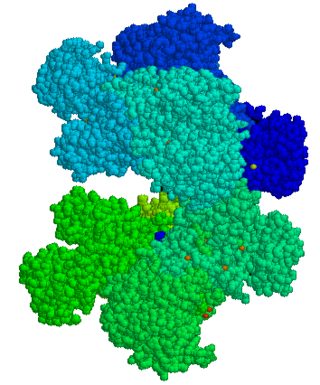
Glucose-6-phosphate dehydrogenase deficiency (G6PDD), also known as favism, is the most common enzyme deficiency worldwide. It is an inborn error of metabolism that predisposes to red blood cell breakdown. Most of the time, those who are affected have no symptoms. Following a specific trigger, symptoms such as yellowish skin, dark urine, shortness of breath, and feeling tired may develop. Complications can include anemia and newborn jaundice. Some people never have symptoms.

Wolfram syndrome, also called DIDMOAD, is a rare autosomal-recessive genetic disorder that causes childhood-onset diabetes mellitus, optic atrophy, and deafness as well as various other possible disorders including neurodegeneration.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found, but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily. While the majority of patients typically exhibit symptoms between the ages of 3 and 12 months, instances of adult onset have also been documented.

The insulin receptor (IR) is a transmembrane receptor that is activated by insulin, IGF-I, IGF-II and belongs to the large class of receptor tyrosine kinase. Metabolically, the insulin receptor plays a key role in the regulation of glucose homeostasis; a functional process that under degenerate conditions may result in a range of clinical manifestations including diabetes and cancer. Insulin signalling controls access to blood glucose in body cells. When insulin falls, especially in those with high insulin sensitivity, body cells begin only to have access to lipids that do not require transport across the membrane. So, in this way, insulin is the key regulator of fat metabolism as well. Biochemically, the insulin receptor is encoded by a single gene INSR, from which alternate splicing during transcription results in either IR-A or IR-B isoforms. Downstream post-translational events of either isoform result in the formation of a proteolytically cleaved α and β subunit, which upon combination are ultimately capable of homo or hetero-dimerisation to produce the ≈320 kDa disulfide-linked transmembrane insulin receptor.

Insulin-like growth factor 1 (IGF-1), also called somatomedin C, is a hormone similar in molecular structure to insulin which plays an important role in childhood growth, and has anabolic effects in adults.

Bloom syndrome is a rare autosomal recessive genetic disorder characterized by short stature, predisposition to the development of cancer, and genomic instability. BS is caused by mutations in the BLM gene which is a member of the RecQ DNA helicase family. Mutations in genes encoding other members of this family, namely WRN and RECQL4, are associated with the clinical entities Werner syndrome and Rothmund–Thomson syndrome, respectively. More broadly, Bloom syndrome is a member of a class of clinical entities that are characterized by chromosomal instability, genomic instability, or both and by cancer predisposition.

Aceruloplasminemia is a rare autosomal recessive disorder in which the liver can not synthesize the protein ceruloplasmin properly, which is needed to transport copper around the blood. Copper deficiency in the brain results in neurological problems that generally appear in adulthood and worsen over time. .
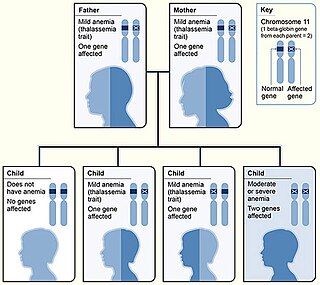
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.

Laron syndrome (LS), also known as growth hormone insensitivity or growth hormone receptor deficiency (GHRD), is an autosomal recessive disorder characterized by a lack of insulin-like growth factor 1 production in response to growth hormone. It is usually caused by inherited growth hormone receptor (GHR) mutations.
Rabson–Mendenhall syndrome is a rare autosomal recessive disorder characterized by severe insulin resistance. The disorder is caused by mutations in the insulin receptor gene. Symptoms include growth abnormalities of the head, face and nails, along with the development of acanthosis nigricans. Treatment involves controlling blood glucose levels by using insulin and incorporating a strategically planned, controlled diet. Also, direct actions against other symptoms may be taken This syndrome usually affects children and has a prognosis of 1–2 years.
3-M syndrome or 3M3 is a rare hereditary disorder characterized by severe growth retardation, facial dysmorphia, and skeletal abnormalities. The name 3-M is derived from the initials of the three researchers who first identified it: Miller, McKusick, and Malvaux and report their findings in the medical literature in 1972. Mutations in any one of the following three genes: CUL7, OBSL1, and CCDC8 are responsible for the occurrence of this disorder. It is inherited through an autosomal recessive pattern and considered very rare, so far less than 100 cases worldwide have been identified. Diagnosis is based on the presence of clinical features. Genetic testing can confirm the diagnosis and identify the specific gene involved. Treatment is aimed at addressing the growth and skeletal problems and may include surgical bone lengthening, adaptive aids, and physical therapy. An endocrinologist may assist with growth hormone replacement and appropriate evaluations during puberty.
Congenital generalized lipodystrophy is an extremely rare autosomal recessive condition, characterized by an extreme scarcity of fat in the subcutaneous tissues. It is a type of lipodystrophy disorder where the magnitude of fat loss determines the severity of metabolic complications. Only 250 cases of the condition have been reported, and it is estimated that it occurs in 1 in 10 million people worldwide.

Wolcott–Rallison syndrome,WRS, is a rare, autosomal recessive disorder with infancy-onset diabetes mellitus, multiple epiphyseal dysplasia, osteopenia, mental retardation or developmental delay, and hepatic and renal dysfunction as main clinical findings. Patients with WRS have mutations in the EIF2AK3 gene, which encodes the eukaryotic translation initiation factor 2-alpha kinase 3. Other disease names include multiple epiphyseal dysplasia and early-onset diabetes mellitus. Most patients with this disease do not survive to adulthood. The majority of WRS patients die from fulminant hepatitis during childhood. There are few reported cases for this disease. Of the 54 families worldwide with reported WRS cases, 22.2% of them are from the Kingdom of Saudi Arabia. Of the 23 WRS patients in Saudi Arabia, all but one is the result of consanguineous marriages. Another country where WRS cases have been found is Kosovo. Here, the Albanian population is also known for consanguineous marriages, but there were some cases involving patients from non-consanguineous parents that were carriers for the same mutant allele.
MODY 2 or GCK-MODY is a form of maturity-onset diabetes of the young. It is due to any of several mutations in the GCK gene on human chromosome 7 for glucokinase. Glucokinase serves as the glucose sensor for the pancreatic beta cell. Normal glucokinase triggers insulin secretion as the glucose exceeds about 90 mg/dl. These loss-of-function mutations result in a glucokinase molecule that is less sensitive or less responsive to rising levels of glucose. The beta cells in MODY 2 have a normal ability to make and secrete insulin, but do so only above an abnormally high threshold. This produces a chronic, mild increase in blood sugar, which is usually asymptomatic. It is usually detected by accidental discovery of mildly elevated blood sugar. An oral glucose tolerance test is much less abnormal than would be expected from the impaired (elevated) fasting blood sugar, since insulin secretion is usually normal once the glucose has exceeded the threshold for that specific variant of the glucokinase enzyme.

Neonatal diabetes mellitus (NDM) is a disease that affects an infant and their body's ability to produce or use insulin.NDM is a kind of diabetes that is monogenic and arises in the first 6 months of life. Infants do not produce enough insulin, leading to an increase in glucose accumulation. It is a rare disease, occurring in only one in 100,000 to 500,000 live births. NDM can be mistaken for the much more common type 1 diabetes, but type 1 diabetes usually occurs later than the first 6 months of life. There are two types of NDM: permanent neonatal diabetes mellitus (PNDM), a lifelong condition, and transient neonatal diabetes mellitus (TNDM), a form of diabetes that disappears during the infant stage but may reappear later in life.
Ichthyosis prematurity syndrome (IPS) is a dermatological disease with known genetic causes. This syndrome is a rare subcategory of autosomal recessive congenital ichthyosis (ARCI). It is associated with complications in the mid-trimester of a pregnancy leading to premature births. Although most prevalent in individuals of Scandinavian origin, there have also been scattered cases in people of Japanese, Italian and Indian ethnicity. This disorder is also referred to as ichthyosis congenital type IV.
Clonal hypereosinophilia, also termed primary hypereosinophilia or clonal eosinophilia, is a grouping of hematological disorders all of which are characterized by the development and growth of a pre-malignant or malignant population of eosinophils, a type of white blood cell that occupies the bone marrow, blood, and other tissues. This population consists of a clone of eosinophils, i.e. a group of genetically identical eosinophils derived from a sufficiently mutated ancestor cell.
WNT4 deficiency is a rare genetic disorder that affects females and it results in the underdevelopment and sometimes absence of the uterus and vagina. WNT4 deficiency is caused by mutations of the WNT4 gene. Abnormally high androgen levels are found in the blood and can initiate and promote the development of male sex characteristics. This is seen as male pattern of hair growth on the chest and face. Those with this genetic defect develop breasts but do not have their period. Mayer–Rokitansky–Küster–Hauser syndrome is a related but distinct syndrome. Some women who have an initial diagnosis of MRKH have later been found to have WNT4 deficiency. Most women with MRKH syndrome do not have genetic mutations of the WNT4 gene. The failure to begin the menstrual cycle may be the initial clinical sign of WNT4 deficiency. WNT4 deficiency can cause significant psychological challenges and counseling is recommended.



