
An autosome is a chromosome that is not a sex chromosome. The members of an autosome pair in a diploid cell have the same morphology, unlike those in allosome pairs which may have different structures. The DNA in autosomes is collectively known as atDNA or auDNA.

A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Cenani–Lenz syndactylism, also known as Cenani–Lenz syndrome or Cenani–syndactylism, is an autosomal recessive congenital malformation syndrome involving both upper and lower extremities.

Tietz syndrome, also called Tietz albinism-deafness syndrome or albinism and deafness of Tietz, is an autosomal dominant congenital disorder characterized by deafness and leucism. It is caused by a mutation in the microphthalmia-associated transcription factor (MITF) gene. Tietz syndrome was first described in 1963 by Walter Tietz (1927–2003) a German Physician working in California.
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections.

Salla disease (SD), is an autosomal recessive lysosomal storage disease characterized by early physical impairment and intellectual disability. It was first described in 1979, after Salla, a municipality in Finnish Lapland. Salla disease is one of 40 Finnish heritage diseases and affects approximately 130 individuals, mainly from Finland and Sweden.
Spondyloperipheral dysplasia is an autosomal dominant disorder of bone growth. The condition is characterized by flattened bones of the spine (platyspondyly) and unusually short fingers and toes (brachydactyly). Some affected individuals also have other skeletal abnormalities, short stature, nearsightedness (myopia), hearing loss, and mental retardation. Spondyloperipheral dysplasia is a subtype of collagenopathy, types II and XI.
Nijmegen breakage syndrome (NBS), is a rare autosomal recessive congenital disorder causing chromosomal instability, probably as a result of a defect in the double Holliday junction DNA repair mechanism and/or the synthesis dependent strand annealing mechanism for repairing double strand breaks in DNA.
Autosomal recessive multiple epiphyseal dysplasia (ARMED), also called epiphyseal dysplasia, multiple, 4 (EDM4), multiple epiphyseal dysplasia with clubfoot or –with bilayered patellae, is an autosomal recessive congenital disorder affecting cartilage and bone development. The disorder has relatively mild signs and symptoms, including joint pain, scoliosis, and malformations of the hands, feet, and knees.

Arakawa's syndrome II is an autosomal dominant metabolic disorder that causes a deficiency of the enzyme tetrahydrofolate-methyltransferase; affected individuals cannot properly metabolize methylcobalamin, a type of Vitamin B12.
Papillon–Lefèvre syndrome (PLS), also known as palmoplantar keratoderma with periodontitis, is an autosomal recessive genetic disorder caused by a deficiency in cathepsin C.
Cerebrotendinous xanthomatosis also called cerebral cholesterosis, is an autosomal recessive form of xanthomatosis. It falls within a group of genetic disorders called the leukodystrophies.
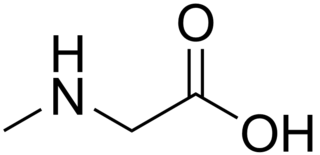
Sarcosinemia (SAR), also called hypersarcosinemia and SARDH deficiency, is a rare autosomal recessive metabolic disorder characterized by an increased concentration of sarcosine in blood plasma and urine ("sarcosinuria"). It can result from an inborn error of sarcosine metabolism, or from severe folate deficiency related to the folate requirement for the conversion of sarcosine to glycine. It is thought to be a relatively benign condition.
White sponge nevus WSN, is an autosomal dominant condition of the oral mucosa. It is caused by a mutations in certain genes coding for keratin, which causes a defect in the normal process of keratinization of the mucosa. This results in lesions which are thick, white and velvety on the inside of the cheeks within the mouth. Usually, these lesions are present from birth or develop during childhood. The condition is entirely harmless, and no treatment is required.
Branchio-oto-renal syndrome (BOR), is an autosomal dominant genetic disorder involving the kidneys, ears, and neck. It often has also been described as Melnick-Fraser syndrome.
Naegeli–Franceschetti–Jadassohn syndrome (NFJS), also known as chromatophore nevus of Naegeli and Naegeli syndrome, is a rare autosomal dominant form of ectodermal dysplasia, characterized by reticular skin pigmentation, diminished function of the sweat glands, the absence of teeth and hyperkeratosis of the palms and soles. One of the most striking features is the absence of fingerprint lines on the fingers.

Rothmund–Thomson syndrome (RTS), is a rare autosomal recessive skin condition.
Woodhouse–Sakati syndrome, is a rare autosomal recessive multisystem disorder which causes malformations throughout the body, and deficiencies affecting the endocrine system.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency. is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
Parastremmatic dwarfism is a rare bone disease that features severe dwarfism, thoracic kyphosis, a distortion and twisting of the limbs, contractures of the large joints, malformations of the vertebrae and pelvis, and incontinence. The disease was first reported in 1970 by Leonard Langer and associates; they used the term parastremmatic from the Greek parastremma, or distorted limbs, to describe it. On X-rays, the disease is distinguished by a "flocky" or lace-like appearance to the bones. The disease is congenital, which means it is apparent at birth. It is caused by a mutation in the TRPV4 gene, located on chromosome 12 in humans. The disease is inherited in an autosomal dominant manner.
