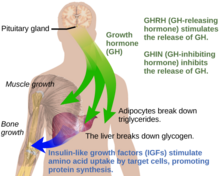
Growth hormone (GH) or somatotropin, also known as human growth hormone in its human form, is a peptide hormone that stimulates growth, cell reproduction, and cell regeneration in humans and other animals. It is thus important in human development. GH also stimulates production of Insulin-like growth factor 1 (IGF-1) and increases the concentration of glucose and free fatty acids. It is a type of mitogen which is specific only to the receptors on certain types of cells. GH is a 191-amino acid, single-chain polypeptide that is synthesized, stored and secreted by somatotropic cells within the lateral wings of the anterior pituitary gland.

Gigantism, also known as giantism, is a condition characterized by excessive growth and height significantly above average. In humans, this condition is caused by over-production of growth hormone in childhood.
Growth hormone deficiency (GHD), or human growth hormone deficiency, is a medical condition resulting from not enough growth hormone (GH). Generally the most noticeable symptom is that an individual attains a short height. Newborns may also present low blood sugar or a small penis size. In adults there may be decreased muscle mass, high cholesterol levels, or poor bone density.

Insulin-like growth factor 1 (IGF-1), also called somatomedin C, is a hormone similar in molecular structure to insulin which plays an important role in childhood growth, and has anabolic effects in adults. In the 1950s IGF-1 was called "sulfation factor" because it stimulated sulfation of cartilage in vitro, and in the 1970s due to its effects it was termed "nonsuppressible insulin-like activity" (NSILA).
Idiopathic short stature (ISS) refers to extreme short stature that does not have a diagnostic explanation after an ordinary growth evaluation. The term has been in use since at least 1975 without a precise percentile or statistical definition of "extreme".

Pituitary adenomas are tumors that occur in the pituitary gland. Most pituitary tumors are benign, approximately 35% are invasive and just 0.1% to 0.2% are carcinomas. Pituitary adenomas represent from 10% to 25% of all intracranial neoplasms and the estimated prevalence rate in the general population is approximately 17%.

Zvi Laron is an Israeli paediatric endocrinologist. Born in Cernăuţi, Romania, Laron is a professor emeritus at Tel Aviv University. In 1966, he described the type of dwarfism later called Laron syndrome. His research opened the way to the treatment of many cases of growth hormone disorders. He was the first to introduce the multidisciplinary treatment for juvenile diabetes.

Growth hormone receptor is a protein that in humans is encoded by the GHR gene. GHR orthologs have been identified in most mammals.
Growth hormone-binding protein (GHBP) is a soluble carrier protein for growth hormone (GH). The full range of functions of GHBP remains to be determined however, current research suggests that the protein is associated with regulation of the GH availability and half-life in the circulatory system, as well as modulating GH receptor function.

The growth-hormone-releasing hormone receptor (GHRHR) is a G-protein-coupled receptor that binds growth hormone-releasing hormone. The GHRHR activates a Gs protein that causes a cascade of cAMP via adenylate cyclase. GHRHR is distinct from the growth hormone secretagogue receptor, where growth hormone releasing peptides act to release growth hormone.
Pegvisomant, sold under the brand name Somavert, is a growth hormone receptor antagonist used in the treatment of acromegaly. It is primarily used if the pituitary gland tumor causing the acromegaly cannot be controlled with surgery or radiation, and the use of somatostatin analogues is unsuccessful, but is also effective as a monotherapy. It is delivered as a powder that is mixed with water and injected under the skin.
Mecasermin, sold under the brand name Increlex, also known as recombinant human insulin-like growth factor-1 (rhIGF-1), is a recombinant form of human insulin-like growth factor 1 (IGF-I) which is used in the long-term treatment of growth failure and short stature in children with severe primary IGF-I deficiency, for instance due to growth hormone deficiency or Laron syndrome.

Aromatase deficiency is a rare condition characterized by extremely low levels or complete absence of the enzyme aromatase activity in the body. It is an autosomal recessive disease resulting from various mutations of gene CYP19 (P450arom) which can lead to ambiguous genitalia and delayed puberty in females, continued linear growth into adulthood and osteoporosis in males and virilization in pregnant mothers. As of 2020, fewer than 15 cases have been identified in genetically male individuals and at least 30 cases in genetically female individuals.
Breast development, also known as mammogenesis, is a complex biological process in primates that takes place throughout a female's life.

John Kopchick is a molecular biologist and co-inventor of the drug Somavert (Pegvisomant), which has improved the lives of acromegalic individuals around the world. He is currently the Goll-Ohio Eminent Scholar and Professor of Molecular Biology in the Department of Biomedical Sciences at the Ohio University Heritage College of Osteopathic Medicine. Dr. Kopchick's groundbreaking work in the field of growth hormone has helped shape the study of endocrinology.

Floating–Harbor syndrome, also known as Pelletier–Leisti syndrome, is a rare disease with fewer than 50 cases described in the literature. It is usually diagnosed in early childhood and is characterized by the triad of proportionate short stature with delayed bone age, characteristic facial appearance, and delayed speech development. Although its cause is unknown, it is thought to result from genetic mutation, and diagnosis is established by the presence of a heterozygous SRCAP mutation in those with clinical findings of FHS.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.

Kowarski syndrome describes cases of growth failure, despite the presence of normal or slightly high blood growth hormone by radioimmunoassay (RIA-GH) and low serum IGF1, and who exhibit a significant increase in growth rate following recombinant GH therapy.
Acromesomelic dysplasia is a rare skeletal disorder that causes abnormal bone and cartilage development, leading to shortening of the forearms, lower legs, hands, feet, fingers, and toes. Five different genetic mutations have been implicated in the disorder. Treatment is individualized but is generally aimed at palliating symptoms, for example, treatment of kyphosis and lumbar hyperlordosis.

The hypothalamic–pituitary–somatotropic axis, or hypothalamic–pituitary–somatic axis, also known as the hypothalamic–pituitary–growth axis, is a hypothalamic–pituitary axis which includes the secretion of growth hormone from the somatotropes of the pituitary gland into the circulation and the subsequent stimulation of insulin-like growth factor 1 production by GH in tissues such as, namely, the liver. Other hypothalamic–pituitary hormones such as growth hormone-releasing hormone, growth hormone-inhibiting hormone, and ghrelin (GHS) are involved in the control of GH secretion from the pituitary gland. The HPS axis is involved in postnatal human growth. Individuals with growth hormone deficiency or Laron syndrome show symptoms like short stature, dwarfism and obesity, but are also protected from some forms of cancer. Conversely, acromegaly and gigantism are conditions of GH and IGF-1 excess usually due to a pituitary tumor, and are characterized by overgrowth and tall stature.