
Colorectal cancer (CRC), also known as bowel cancer, colon cancer, or rectal cancer, is the development of cancer from the colon or rectum. Signs and symptoms may include blood in the stool, a change in bowel movements, weight loss, and fatigue. Most colorectal cancers are due to old age and lifestyle factors, with only a small number of cases due to underlying genetic disorders. Risk factors include diet, obesity, smoking, and lack of physical activity. Dietary factors that increase the risk include red meat, processed meat, and alcohol. Another risk factor is inflammatory bowel disease, which includes Crohn's disease and ulcerative colitis. Some of the inherited genetic disorders that can cause colorectal cancer include familial adenomatous polyposis and hereditary non-polyposis colon cancer; however, these represent less than 5% of cases. It typically starts as a benign tumor, often in the form of a polyp, which over time becomes cancerous.

In anatomy, a polyp is an abnormal growth of tissue projecting from a mucous membrane. If it is attached to the surface by a narrow elongated stalk, it is said to be pedunculated; if it is attached without a stalk, it is said to be sessile. Polyps are commonly found in the colon, stomach, nose, ear, sinus(es), urinary bladder, and uterus. They may also occur elsewhere in the body where there are mucous membranes, including the cervix, vocal folds, and small intestine. Some polyps are tumors (neoplasms) and others are non-neoplastic, for example hyperplastic or dysplastic, which are benign. The neoplastic ones are usually benign, although some can be pre-malignant, or concurrent with a malignancy.

Gardner's syndrome is a subtype of familial adenomatous polyposis (FAP). Gardner syndrome is an autosomal dominant form of polyposis characterized by the presence of multiple polyps in the colon together with tumors outside the colon. The extracolonic tumors may include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas, as well as the occurrence of desmoid tumors in approximately 15% of affected individuals.
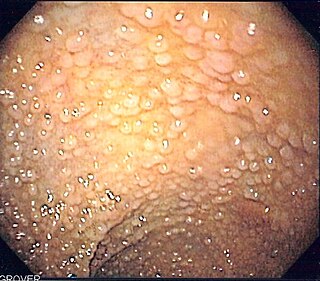
Familial adenomatous polyposis (FAP) is an autosomal dominant inherited condition in which numerous adenomatous polyps form mainly in the epithelium of the large intestine. While these polyps start out benign, malignant transformation into colon cancer occurs when they are left untreated. Three variants are known to exist, FAP and attenuated FAP are caused by APC gene defects on chromosome 5 while autosomal recessive FAP is caused by defects in the MUTYH gene on chromosome 1. Of the three, FAP itself is the most severe and most common; although for all three, the resulting colonic polyps and cancers are initially confined to the colon wall. Detection and removal before metastasis outside the colon can greatly reduce and in many cases eliminate the spread of cancer.

Peutz–Jeghers syndrome is an autosomal dominant genetic disorder characterized by the development of benign hamartomatous polyps in the gastrointestinal tract and hyperpigmented macules on the lips and oral mucosa (melanosis). This syndrome can be classed as one of various hereditary intestinal polyposis syndromes and one of various hamartomatous polyposis syndromes. It has an incidence of approximately 1 in 25,000 to 300,000 births.

A benign tumor is a mass of cells (tumor) that does not invade neighboring tissue or metastasize. Compared to malignant (cancerous) tumors, benign tumors generally have a slower growth rate. Benign tumors have relatively well differentiated cells. They are often surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.

Mismatch repair cancer syndrome (MMRCS) is a cancer syndrome associated with biallelic DNA mismatch repair mutations. It is also known as Turcot syndrome and by several other names.

Cowden syndrome is an autosomal dominant inherited condition characterized by benign overgrowths called hamartomas as well as an increased lifetime risk of breast, thyroid, uterine, and other cancers. It is often underdiagnosed due to variability in disease presentation, but 99% of patients report mucocutaneous symptoms by age 20–29. Despite some considering it a primarily dermatologic condition, Cowden's syndrome is a multi-system disorder that also includes neurodevelopmental disorders such as macrocephaly.

Bannayan–Riley–Ruvalcaba syndrome (BRRS) is a rare overgrowth syndrome and hamartomatous disorder with occurrence of multiple subcutaneous lipomas, macrocephaly and hemangiomas. The disease is inherited in an autosomal dominant manner. The disease belongs to a family of hamartomatous polyposis syndromes, which also includes Peutz–Jeghers syndrome, juvenile polyposis and Cowden syndrome. Mutation of the PTEN gene underlies this syndrome, as well as Cowden syndrome, Proteus syndrome, and Proteus-like syndrome, these four syndromes are referred to as PTEN Hamartoma-Tumor Syndromes.
Pallister–Hall syndrome (PHS) is a rare genetic disorder that affects various body systems. The main features are a non-cancerous mass on the hypothalamus and extra digits (polydactylism). The prevalence of Pallister-Hall Syndrome is unknown; about 100 cases have been reported in publication.

Fundic gland polyposis is a medical syndrome where the fundus and the body of the stomach develop many fundic gland polyps. The condition has been described both in patients with familial adenomatous polyposis (FAP) and attenuated variants (AFAP), and in patients in whom it occurs sporadically.
Cronkhite–Canada syndrome is a rare syndrome characterized by multiple polyps of the digestive tract. It is sporadic, and it is currently considered acquired and idiopathic.

Hereditary breast–ovarian cancer syndromes (HBOC) are cancer syndromes that produce higher than normal levels of breast cancer, ovarian cancer and additional cancers in genetically related families. It accounts for 90% of the hereditary cancers. The hereditary factors may be proven or suspected to cause the pattern of breast and ovarian cancer occurrences in the family. The name HBOC may be misleading because it implies that this genetic susceptibility to cancer is mainly in women. In reality, both sexes have the same rates of gene mutations and HBOC can predispose to other cancers including prostate cancer and pancreatic cancer. For this reason, the term "King syndrome" has recently come into use. The new name references Mary-Claire King who identified the genes BRCA1 and BRCA2.
A colorectal polyp is a polyp occurring on the lining of the colon or rectum. Untreated colorectal polyps can develop into colorectal cancer.
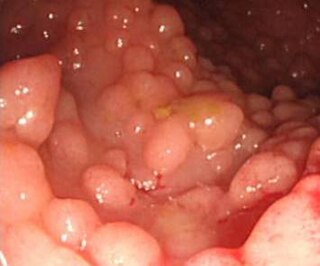
A hereditary cancer syndrome is a genetic disorder in which inherited genetic mutations in one or more genes predispose the affected individuals to the development of cancer and may also cause early onset of these cancers. Hereditary cancer syndromes often show not only a high lifetime risk of developing cancer, but also the development of multiple independent primary tumors.
MUTYH-associated polyposis is an autosomal recessive polyposis syndrome. The disorder is caused by mutations in both alleles of the DNA repair gene, MUTYH. The MUTYH gene encodes a base excision repair protein, which corrects oxidative damage to DNA. Affected individuals have an increased risk of colorectal cancer, precancerous colon polyps (adenomas) and an increased risk of several additional cancers. About 1–2 percent of the population possess a mutated copy of the MUTYH gene, and less than 1 percent of people have the MUTYH associated polyposis syndrome. The presence of 10 or more colon adenomas should prompt consideration of MUTYH-associated polyposis, familial adenomatous polyposis and similar syndromes.
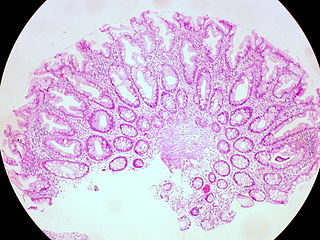
Serrated polyposis syndrome (SPS), previously known as hyperplastic polyposis syndrome, is a disorder characterized by the appearance of serrated polyps in the colon. While serrated polyposis syndrome does not cause symptoms, the condition is associated with a higher risk of colorectal cancer (CRC). The lifelong risk of CRC is between 25 and 40%. SPS is the most common polyposis syndrome affecting the colon, but is under recognized due to a lack of systemic long term monitoring. Diagnosis requires colonoscopy, and is defined by the presence of either of two criteria: ≥5 serrated lesions/polyps proximal to the rectum, or >20 serrated lesions/polyps of any size distributed throughout the colon with 5 proximal to the rectum.
Polymerase proofreading-associated polyposis (PPAP) is an autosomal dominant hereditary cancer syndrome, which is characterized by numerous polyps in the colon and an increased risk of colorectal cancer. It is caused by germline mutations in DNA polymerase ε (POLE) and δ (POLD1). Affected individuals develop numerous polyps called colorectal adenomas. Compared with other polyposis syndromes, Polymerase proofreading-associated polyposis is rare. Genetic testing can help exclude similar syndromes, such as Familial adenomatous polyposis and MUTYH-associated polyposis. Endometrial cancer, duodenal polyps and duodenal cancer may also occur.
Juvenile polyps are a type of polyp found in the colon. While juvenile polyps are typically found in children, they may be found in people of any age. Juvenile polyps are a type of hamartomatous polyps, which consist of a disorganized mass of tissue. They occur in about two percent of children. Juvenile polyps often do not cause symptoms (asymptomatic); when present, symptoms usually include gastrointestinal bleeding and prolapse through the rectum. Removal of the polyp (polypectomy) is warranted when symptoms are present, for treatment and definite histopathological diagnosis. In the absence of symptoms, removal is not necessary. Recurrence of polyps following removal is relatively common. Juvenile polyps are usually sporadic, occurring in isolation, although they may occur as a part of juvenile polyposis syndrome. Sporadic juvenile polyps may occur in any part of the colon, but are usually found in the distal colon. In contrast to other types of colon polyps, juvenile polyps are not premalignant and are not usually associated with a higher risk of cancer; however, individuals with juvenile polyposis syndrome are at increased risk of gastric and colorectal cancer., Unlike juvenile polyposis syndrome, solitary juvenile polyps do not require follow up with surveillance colonoscopy.
Gardner fibroma (GF) is a benign fibroblastic tumor. GF tumors typically develop in the dermis and adjacent subcutaneous tissue lying just below the dermis. These tumors typically occur on the back, abdomen, and other superficial sites but in rare cases have been diagnoses in internal sites such as the retroperitoneum and around the large blood vessels in the upper thoracic cavity. The World Health Organization, 2020, classified Gardner fibroma as a benign tumor in the category of fibroblastic and myofibroblastic tumors.
