
CHARGE syndrome is a rare syndrome caused by a genetic disorder. First described in 1979, the acronym "CHARGE" came into use for newborn children with the congenital features of coloboma of the eye, heart defects, atresia of the nasal choanae, restricted growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness. These features are no longer used in making a diagnosis of CHARGE syndrome, but the name remains. About two thirds of cases are due to a CHD7 mutation. CHARGE syndrome occurs only in 0.1–1.2 per 10,000 live births; as of 2009, it was the leading cause of congenital deafblindness in the US.

The VACTERL association refers to a recognized group of birth defects which tend to co-occur. This pattern is a recognized association, as opposed to a syndrome, because there is no known pathogenetic cause to explain the grouped incidence.
Adams–Oliver syndrome (AOS) is a rare congenital disorder characterized by defects of the scalp and cranium, transverse defects of the limbs, and mottling of the skin.

Nijmegen breakage syndrome (NBS) is a rare autosomal recessive congenital disorder causing chromosomal instability, probably as a result of a defect in the double Holliday junction DNA repair mechanism and/or the synthesis dependent strand annealing mechanism for repairing double strand breaks in DNA.
Fumarase deficiency is an exceedingly rare autosomal recessive metabolic disorder in the Krebs cycle, characterized by a deficiency of the enzyme fumarate hydratase, which causes a buildup of fumaric acid in the urine and a deficiency of malate. Only 13 cases were known worldwide in 1990, after which a cluster of 20 cases was documented in a community in Arizona, US that has practiced successive endogamy.
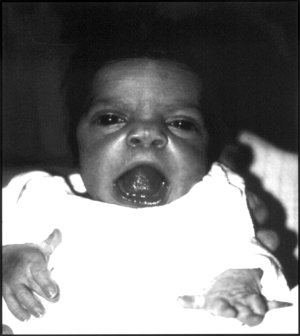
Robinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow, along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.

Hay–Wells syndrome is one of at least 150 known types of ectodermal dysplasia. These disorders affect tissues that arise from the ectodermal germ layer, such as skin, hair, and nails.
Dubowitz syndrome is a rare genetic disorder characterized by microcephaly, stunted growth, and a receding chin. Symptoms vary among patients, but other characteristics include a soft, high-pitched voice, partial webbing of the fingers and toes, palate deformations, genital abnormalities, language difficulties, and an aversion to crowds. The pathogenesis of the disease is yet to be identified, and no medical tests can definitively diagnose the disease. The primary method of diagnosis is to identify facial phenotypes. Since it was first described in 1965 by English physician Victor Dubowitz, over 140 cases have been reported worldwide. Although the majority of cases have been reported from the United States, Germany, and Russia, the disorder appears to affect both genders and all ethnicities equally.

Aristaless related homeobox is a protein that in humans is encoded by the ARX gene.

Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.
Boomerang dysplasia is a lethal form of osteochondrodysplasia known for a characteristic congenital feature in which bones of the arms and legs are malformed into the shape of a boomerang. Death usually occurs in early infancy due to complications arising from overwhelming systemic bone malformations.
EEM syndrome is an autosomal recessive congenital malformation disorder affecting tissues associated with the ectoderm, and also the hands, feet and eyes.
Scalp–ear–nipple (SEN) syndrome is a condition associated with aplasia cutis congenita.
Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.

Distal 18q- is a genetic condition caused by a deletion of genetic material within one of the two copies of chromosome 18. The deletion involves the distal section of 18q and typically extends to the tip of the long arm of chromosome 18.

Gómez–López-Hernández syndrome (GLH) or cerebellotrigeminal-dermal dysplasia is a rare neurocutaneous (Phakomatosis) disorder affecting the trigeminal nerve and causing several other neural and physical abnormalities. Gómez–López-Hernández syndrome has been diagnosed in only 34 people. Cases of Gómez–López-Hernández syndrome may be under-reported as other diseases share the characteristics of cerebellar malformation shown in Gómez–López-Hernández syndrome. Gómez–López-Hernández syndrome was first characterized in 1979.

Ankyrin repeat domain 11 is a protein that in humans is encoded by the ANKRD11 gene.

Strømme syndrome is a very rare autosomal recessive genetic condition characterised by intestinal atresia, eye abnormalities and microcephaly. The intestinal atresia is of the "apple-peel" type, in which the remaining intestine is twisted around its main artery. The front third of the eye is typically underdeveloped, and there is usually moderate developmental delay. Less common features include an atrial septal defect, increased muscle tone or skeletal abnormalities. Physical features may include short stature, large, low-set ears, a small jaw, a large mouth, epicanthic folds, or fine, sparse hair.
XK aprosencephaly is an extremely rare congenital disorder characterized by the absence of the embryonic forebrain. Because the prosencephalon gives way to the cerebral cortex, survival with aprosencephaly is not possible outside utero. The external symptoms are similar to holoprosencephaly, a related disorder, including a smaller than normal head (microcephaly), small eyeballs (microphthalmia), a small mouth (microstomia), anal atresia, and abnormalities of the external genitalia, radius, nostrils, and pharynx (throat).
Mandibulofacial dysostosis with microcephaly syndrome, also known as growth delay-intellectual disability-mandibulofacial dysostosis-microcephaly-cleft palate syndrome, mandibulofacial dysostosis, guion-almeida type, or simply as MFDM syndrome is a rare genetic disorder which is characterized by developmental delays, intellectual disabilities, and craniofacial dysmorphisms.
