Related Research Articles
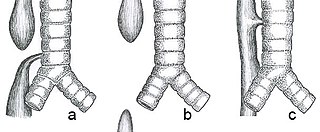
Esophageal atresia is a congenital medical condition that affects the alimentary tract. It causes the esophagus to end in a blind-ended pouch rather than connecting normally to the stomach. It comprises a variety of congenital anatomic defects that are caused by an abnormal embryological development of the esophagus. It is characterized anatomically by a congenital obstruction of the esophagus with interruption of the continuity of the esophageal wall.

Anotia describes a rare congenital deformity that involves the complete absence of the pinna, the outer projected portion of the ear, and narrowing or absence of the ear canal. This contrasts with microtia, in which a small part of the pinna is present. Anotia and microtia may occur unilaterally or bilaterally. This deformity results in conductive hearing loss, deafness.
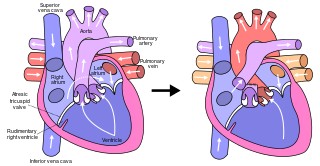
The Fontan procedure or Fontan–Kreutzer procedure is a palliative surgical procedure used in children with univentricular hearts. It involves diverting the venous blood from the inferior vena cava (IVC) and superior vena cava (SVC) to the pulmonary arteries. The procedure varies for differing congenital heart pathologies. For example in tricuspid atresia, the procedure can be done where the blood does not pass through the morphologic right ventricle; i.e., the systemic and pulmonary circulations are placed in series with the functional single ventricle. Whereas in hypoplastic left heart syndrome, the heart is more reliant on the more functional right ventricle to provide blood flow to the systemic circulation. The procedure was initially performed in 1968 by Francis Fontan and Eugene Baudet from Bordeaux, France, published in 1971, simultaneously described in 1971 by Guillermo Kreutzer from Buenos Aires, Argentina, and finally published in 1973.
Situs ambiguus is a rare congenital defect in which the major visceral organs are distributed abnormally within the chest and abdomen. Clinically heterotaxy spectrum generally refers to any defect of Left-right asymmetry and arrangement of the visceral organs; however, classical heterotaxy requires multiple organs to be affected. This does not include the congenital defect situs inversus, which results when arrangement of all the organs in the abdomen and chest are mirrored, so the positions are opposite the normal placement. Situs inversus is the mirror image of situs solitus, which is normal asymmetric distribution of the abdominothoracic visceral organs. Situs ambiguus can also be subdivided into left-isomerism and right isomerism based on the defects observed in the spleen, lungs and atria of the heart.
Potter sequence is the atypical physical appearance of a baby due to oligohydramnios experienced when in the uterus. It includes clubbed feet, pulmonary hypoplasia and cranial anomalies related to the oligohydramnios. Oligohydramnios is the decrease in amniotic fluid volume sufficient to cause deformations in morphogenesis of the baby.

An imperforate anus or anorectal malformations (ARMs) are birth defects in which the rectum is malformed. ARMs are a spectrum of different congenital anomalies which vary from fairly minor lesions to complex anomalies. The cause of ARMs is unknown; the genetic basis of these anomalies is very complex because of their anatomical variability. In 8% of patients, genetic factors are clearly associated with ARMs. Anorectal malformation in Currarino syndrome represents the only association for which the gene HLXB9 has been identified.

Microtia is a congenital deformity where the auricle is underdeveloped. A completely undeveloped pinna is referred to as anotia. Because microtia and anotia have the same origin, it can be referred to as microtia-anotia. Microtia can be unilateral or bilateral. Microtia occurs in 1 out of about 8,000–10,000 births. In unilateral microtia, the right ear is most commonly affected. It may occur as a complication of taking Accutane (isotretinoin) during pregnancy.

Fraser syndrome is an autosomal recessive congenital disorder, identified by several developmental anomalies. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.

The VACTERL association refers to a recognized group of birth defects which tend to co-occur. This pattern is a recognized association, as opposed to a syndrome, because there is no known pathogenetic cause to explain the grouped incidence.

Goldenhar syndrome is a rare congenital defect characterized by incomplete development of the ear, nose, soft palate, lip and mandible on usually one side of the body. Common clinical manifestations include limbal dermoids, preauricular skin tags and strabismus. It is associated with anomalous development of the first branchial arch and second branchial arch.
Hematocolpos is a medical condition in which the vagina is pooled with menstrual blood due to multiple factors leading to the blockage of menstrual blood flow. The medical definition of hematocolpos is 'an accumulation of blood within the vagina'. It is often caused by the combination of menstruation with an imperforate hymen. It is sometimes seen in Robinow syndrome, uterus didelphys, or other vaginal anomalies.
Tracheal agenesis is a rare birth defect with a prevalence of less than 1 in 50,000 in which the trachea fails to develop, resulting in an impaired communication between the larynx and the alveoli of the lungs. Although the defect is normally fatal, occasional cases have been reported of long-term survival following surgical intervention.

13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:

Vaginal anomalies are abnormal structures that are formed during the prenatal development of the female reproductive system and are rare congenital defects that result in an abnormal or absent vagina.

Strømme syndrome is a very rare autosomal recessive genetic condition characterised by intestinal atresia, eye abnormalities and microcephaly. The intestinal atresia is of the "apple-peel" type, in which the remaining intestine is twisted around its main artery. The front third of the eye is typically underdeveloped, and there is usually moderate developmental delay. Less common features include an atrial septal defect, increased muscle tone or skeletal abnormalities. Physical features may include short stature, large, low-set ears, a small jaw, a large mouth, epicanthic folds, or fine, sparse hair.

Pulmonary agenesis is an inborn lung underdevelopment that is rare and potentially lethal. The disorder is caused by a complete developmental arrest of the primitive lung during embryonic life, and it is often associated with other developmental defects. Bilateral and unilateral pulmonary agenesis are classified, depending on whether one side of the lung or both sides are affected. Bilateral pulmonary agenesis is lethal, while the mortality rate of unilateral pulmonary agenesis is higher than 50%. Depending on the severity, the symptom ranges from none to various respiratory complaints. It is detectable prenatally, however, its nonspecific clinical features act as the obstacle for diagnosing. The exact cause of pulmonary agenesis is still obscure. However, theories have been raised regarding the vascular, iatrogenic, viral and genetic causes of pulmonary agenesis in an attempt to explain the pathogenesis of the disorder. In most cases of pulmonary agenesis, surgical resection is performed to remove the malformed lobe or the entire defected lung of the patient depending on the severity of the respiratory impairment.

Pulmonary atresia with ventricular septal defect is a rare birth defect characterized by pulmonary valve atresia occurring alongside a defect on the right ventricular outflow tract.
References
- ↑ Dorland's illustrated medical dictionary. Dorland, W. A. Newman (William Alexander Newman), 1864-1956. (32nd ed.). Philadelphia, PA: Saunders/Elsevier. 2012. p. 174. ISBN 978-1-4160-6257-8. OCLC 706780870.
{{cite book}}: CS1 maint: others (link) - ↑ "Anotia". Children's Hospital of Philadelphia. July 30, 2014. Retrieved November 14, 2023.
- ↑ Chardot, Christophe (July 26, 2006). "Biliary atresia". Orphanet Journal of Rare Diseases. Springer Science and Business Media LLC. 1 (1): 28. doi: 10.1186/1750-1172-1-28 . ISSN 1750-1172. PMC 1560371 . PMID 16872500.
- ↑ Schreiber, Richard A.; Kleinman, Ronald E. (2002). "Biliary Atresia". Journal of Pediatric Gastroenterology and Nutrition. Ovid Technologies (Wolters Kluwer Health). 35: 11–16. doi: 10.1097/00005176-200207001-00005 . ISSN 0277-2116. PMID 12151815 . Retrieved November 14, 2023.
- ↑ Wang, Yuqi; Dai, Weimin; Sun, Yu'e; Chu, Xiangyang; Yang, Bo; Zhao, Ming (2012). "Congenital Bronchial Atresia: Diagnosis and Treatment". International Journal of Medical Sciences. Ivyspring International Publisher. 9 (3): 207–212. doi: 10.7150/ijms.3690 . ISSN 1449-1907. PMC 3298011 . PMID 22408569.
- ↑ Kwong, Kelvin M. (June 9, 2015). "Current Updates on Choanal Atresia". Frontiers in Pediatrics. Frontiers Media SA. 3: 52. doi: 10.3389/fped.2015.00052 . ISSN 2296-2360. PMC 4460812 . PMID 26106591.
- ↑ Sfeir, R.; Michaud, L.; Salleron, J.; Gottrand, F. (2013). "Epidemiology of esophageal atresia". Diseases of the Esophagus. Oxford University Press (OUP). 26 (4): 354–355. doi: 10.1111/dote.12051 . ISSN 1120-8694. PMID 23679022.
- ↑ Pinheiro, Paulo Fernando Martins (2012). "Current knowledge on esophageal atresia". World Journal of Gastroenterology. Baishideng Publishing Group Inc. 18 (28): 3662–3672. doi: 10.3748/wjg.v18.i28.3662 . ISSN 1007-9327. PMC 3406418 . PMID 22851858.
- ↑ Sfeir, Rony; Bonnard, Arnaud; Khen-Dunlop, Naziha; Auber, Frederic; Gelas, Thomas; Michaud, Laurent; Podevin, Guillaume; Breton, Anne; Fouquet, Virginie; Piolat, Christian; Lemelle, Jean Louis; Petit, Thierry; Lavrand, Frederic; Becmeur, Francis; Polimerol, Marie Laurence; Michel, Jean Luc; Elbaz, Frederic; Habonimana, Eric; Allal, Hassan; Lopez, Emmanuel; Lardy, Hubert; Morineau, Marianne; Pelatan, Cécile; Merrot, Thierry; Delagausie, Pascal; de Vries, Philline; Levard, Guillaume; Buisson, Phillippe; Sapin, Emmanuel; Jaby, Olivier; Borderon, Corinne; Weil, Dominique; Gueiss, Stephane; Aubert, Didier; Echaieb, Anais; Fourcade, Laurent; Breaud, Jean; Laplace, Christophe; Pouzac, Myriam; Duhamel, Alain; Gottrand, Frederic (2013). "Esophageal atresia: Data from a national cohort". Journal of Pediatric Surgery. Elsevier BV. 48 (8): 1664–1669. doi:10.1016/j.jpedsurg.2013.03.075. ISSN 0022-3468. PMID 23932604. S2CID 34736647 . Retrieved November 14, 2023.
- ↑ Höllwarth, Michael E.; Till, Holger (2020). "Esophageal Atresia". Pediatric Surgery. Berlin, Heidelberg: Springer Berlin Heidelberg. pp. 661–680. doi:10.1007/978-3-662-43588-5_48. ISBN 978-3-662-43587-8 . Retrieved November 14, 2023.
- ↑ McGee EA, Horne J (2018). "Follicle Atresia". In Skinner MK (ed.). Encyclopedia of Reproduction (Second ed.). Oxford: Academic Press. pp. 87–91. doi:10.1016/b978-0-12-801238-3.64395-7. ISBN 978-0-12-815145-7.
- ↑ Liu Z, Li F, Xue J, Wang M, Lai S, Bao H, He S (August 2020). "Esculentoside A rescues granulosa cell apoptosis and folliculogenesis in mice with premature ovarian failure". Aging. 12 (17): 16951–16962. doi:10.18632/aging.103609. PMC 7521512 . PMID 32759462.
- ↑ Brantberg, A.; Blaas, H.-G. K.; Haugen, S. E.; Isaksen, C. V.; Eik-Nes, S. H. (November 23, 2006). "Imperforate anus: a relatively common anomaly rarely diagnosed prenatally". Ultrasound in Obstetrics & Gynecology. Wiley. 28 (7): 904–910. doi: 10.1002/uog.3862 . ISSN 0960-7692. PMID 17091530.
- ↑ Subbarayan, Devi (2015). "Histomorphological Features of Intestinal Atresia and its Clinical Correlation". Journal of Clinical and Diagnostic Research. JCDR Research and Publications. 9 (11): EC26–EC29. doi: 10.7860/jcdr/2015/13320.6838 . ISSN 2249-782X. PMC 4668418 . PMID 26674207.
- ↑ Gupta, Shilpi; Gupta, Rahul; Ghosh, Soumyodhriti; Gupta, Arun Kumar; Shukla, Arvind; Chaturvedi, Vinita; Mathur, Praveen (October 7, 2016). "Intestinal Atresia: Experience at a Busy Center of North-West India". Journal of Neonatal Surgery. 5 (4): 51. doi: 10.21699/jns.v5i4.405 . ISSN 2226-0439. PMC 5117274 . PMID 27896159.
- ↑ Online Mendelian Inheritance in Man (OMIM): Microtia-Anotia - 600674
- ↑ Pretest self assessment and review for the USMLE, pediatrics, 12th edition, question 84, general pediatrics
- ↑ Shastry, SrikanthM; Kolte, SachinS; Sanagapati, PandurangaR (2012). "Potter′s sequence". Journal of Clinical Neonatology. Medknow. 1 (3): 157–159. doi: 10.4103/2249-4847.101705 . ISSN 2249-4847. PMC 3762025 . PMID 24027716.
- ↑ Jelin, Angie (2021). "Renal agenesis". American Journal of Obstetrics and Gynecology. Elsevier BV. 225 (5): 28–30. doi: 10.1016/j.ajog.2021.06.048 . ISSN 0002-9378. PMID 34507792 . Retrieved November 14, 2023.
- 1 2 Murthy, Raghav; Nigro, John; Karamlou, Tara (2019-01-01), Ungerleider, Ross M.; Meliones, Jon N.; Nelson McMillan, Kristen; Cooper, David S. (eds.), "65 - Tricuspid Atresia", Critical Heart Disease in Infants and Children (Third Edition), Philadelphia: Elsevier, pp. 765–777.e3, doi:10.1016/b978-1-4557-0760-7.00065-6, ISBN 978-1-4557-0760-7, S2CID 214741527 , retrieved 2020-11-27
- ↑ "Congenital Heart Defects — Facts about Tricuspid Atresia | CDC". 2019-01-22.
- ↑ Saxena, Amulya K (November 9, 2021). "Vaginal Atresia: Practice Essentials, Anatomy, Pathophysiology". Medscape Reference. Retrieved November 14, 2023.