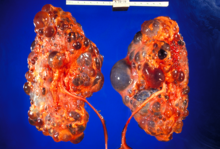
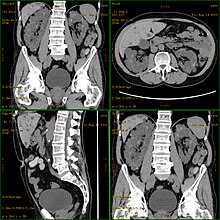
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common, life-threatening inherited human disorders and the most common hereditary kidney disease. It is associated with large interfamilial and intrafamilial variability, which can be explained to a large extent by its genetic heterogeneity and modifier genes. It is also the most common of the inherited cystic kidney diseases — a group of disorders with related but distinct pathogenesis, characterized by the development of renal cysts and various extrarenal manifestations, which in case of ADPKD include cysts in other organs, such as the liver, seminal vesicles, pancreas, and arachnoid membrane, as well as other abnormalities, such as intracranial aneurysms and dolichoectasias, aortic root dilatation and aneurysms, mitral valve prolapse, and abdominal wall hernias. Over 50% of patients with ADPKD eventually develop end stage kidney disease and require dialysis or kidney transplantation. ADPKD is estimated to affect at least one in every 1000 individuals worldwide, making this disease the most common inherited kidney disorder with a diagnosed prevalence of 1:2000 and incidence of 1:3000-1:8000 in a global scale.

The cilium is a membrane-bound organelle found on most types of eukaryotic cell. Cilia are absent in bacteria and archaea. The cilium has the shape of a slender threadlike projection that extends from the surface of the much larger cell body. Eukaryotic flagella found on sperm cells and many protozoans have a similar structure to motile cilia that enables swimming through liquids; they are longer than cilia and have a different undulating motion.

Kidney disease, or renal disease, technically referred to as nephropathy, is damage to or disease of a kidney. Nephritis is an inflammatory kidney disease and has several types according to the location of the inflammation. Inflammation can be diagnosed by blood tests. Nephrosis is non-inflammatory kidney disease. Nephritis and nephrosis can give rise to nephritic syndrome and nephrotic syndrome respectively. Kidney disease usually causes a loss of kidney function to some degree and can result in kidney failure, the complete loss of kidney function. Kidney failure is known as the end-stage of kidney disease, where dialysis or a kidney transplant is the only treatment option.
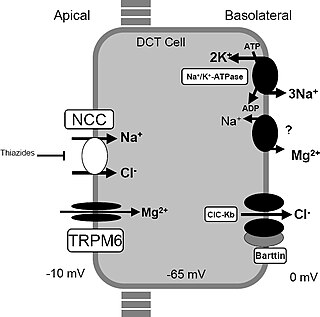
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH. It is the most frequent hereditary salt-losing tubulopathy. Gitelman syndrome is caused by disease-causing variants on both alleles of the SLC12A3 gene. The SLC12A3 gene encodes the thiazide-sensitive sodium-chloride cotransporter, which can be found in the distal convoluted tubule of the kidney.
Secondary hypertension is a type of hypertension which by definition is caused by an identifiable underlying primary cause. It is much less common than the other type, called essential hypertension, affecting only 5-10% of hypertensive patients. It has many different causes including endocrine diseases, kidney diseases, and tumors. It also can be a side effect of many medications.

Cystic kidney disease refers to a wide range of hereditary, developmental, and acquired conditions and with the inclusion of neoplasms with cystic changes, over 40 classifications and subtypes have been identified. Depending on the disease classification, the presentation may be at birth, or much later into adult life. Cystic disease may involve one or both kidneys and may, or may not, occur in the presence of other anomalies. A higher incidence is found in males and prevalence increases with age. Renal cysts have been reported in more than 50% of patients over the age of 50. Typically, cysts grow up to 2.88 mm annually and may cause related pain and/or hemorrhage.

Medullary cystic kidney disease (MCKD) is an autosomal dominant kidney disorder characterized by tubulointerstitial sclerosis leading to end-stage renal disease. Because the presence of cysts is neither an early nor a typical diagnostic feature of the disease, and because at least 4 different gene mutations may give rise to the condition, the name autosomal dominant tubulointerstitial kidney disease (ADTKD) has been proposed, to be appended with the underlying genetic variant for a particular individual. Importantly, if cysts are found in the medullary collecting ducts they can result in a shrunken kidney, unlike that of polycystic kidney disease. There are two known forms of medullary cystic kidney disease, mucin-1 kidney disease 1 (MKD1) and mucin-2 kidney disease/uromodulin kidney disease (MKD2). A third form of the disease occurs due to mutations in the gene encoding renin (ADTKD-REN), and has formerly been known as familial juvenile hyperuricemic nephropathy type 2.
Branchio-oto-renal syndrome (BOR) is an autosomal dominant genetic disorder involving the kidneys, ears, and neck. It is also known as Melnick-Fraser syndrome.
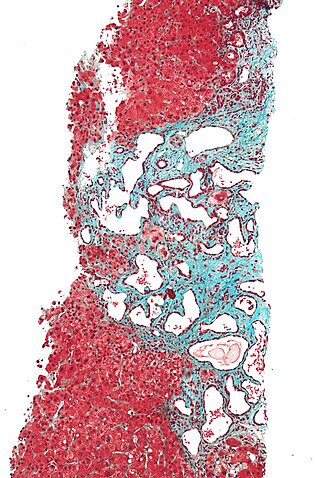
Polycystic liver disease (PLD) usually describes the presence of multiple cysts scattered throughout normal liver tissue. PLD is commonly seen in association with autosomal-dominant polycystic kidney disease, with a prevalence of 1 in 400 to 1000, and accounts for 8–10% of all cases of end-stage renal disease. The much rarer autosomal-dominant polycystic liver disease will progress without any kidney involvement.

Fibrocystin is a large, receptor-like protein that is thought to be involved in the tubulogenesis and/or maintenance of duct-lumen architecture of epithelium. FPC associates with the primary cilia of epithelial cells and co-localizes with the Pkd2 gene product polycystin-2 (PC2), suggesting that these two proteins may function in a common molecular pathway.

Nephronophthisis is a genetic disorder of the kidneys which affects children. It is classified as a medullary cystic kidney disease. The disorder is inherited in an autosomal recessive fashion and, although rare, is the most common genetic cause of childhood kidney failure. It is a form of ciliopathy. Its incidence has been estimated to be 0.9 cases per million people in the United States, and 1 in 50,000 births in Canada.

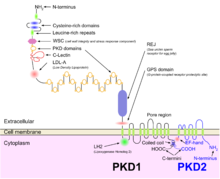
Polycystin 1 (PC1) is a protein that in humans is encoded by the PKD1 gene. Mutations of PKD1 are associated with most cases of autosomal dominant polycystic kidney disease, a severe hereditary disorder of the kidneys characterised by the development of renal cysts and severe kidney dysfunction.

Polycystin-2(PC2) is a protein that in humans is encoded by the PKD2 gene.

A ciliopathy is any genetic disorder that affects the cellular cilia or the cilia anchoring structures, the basal bodies, or ciliary function. Primary cilia are important in guiding the process of development, so abnormal ciliary function while an embryo is developing can lead to a set of malformations that can occur regardless of the particular genetic problem. The similarity of the clinical features of these developmental disorders means that they form a recognizable cluster of syndromes, loosely attributed to abnormal ciliary function and hence called ciliopathies. Regardless of the actual genetic cause, it is clustering of a set of characteristic physiological features which define whether a syndrome is a ciliopathy.
Autosomal recessive polycystic kidney disease (ARPKD) is the recessive form of polycystic kidney disease. It is associated with a group of congenital fibrocystic syndromes. Mutations in the PKHD1 cause ARPKD.

Lixivaptan (VPA-985) is an orally-active, non-peptide, selective vasopressin 2 receptor antagonist being developed as an investigational drug by Palladio Biosciences, Inc. (Palladio), a subsidiary of Centessa Pharmaceuticals plc. As of December 2021, lixivaptan is in Phase III clinical development for the treatment of Autosomal dominant polycystic kidney disease (ADPKD), the most common form of polycystic kidney disease. The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to lixivaptan for the treatment of ADPKD.
Glomerulocystic kidney disease (GCKD) is a cystic disorder of the kidneys. GCKD involves cystic dilation of Bowman's capsule. It can occur with or without congenital abnormality.
The Polycystin Cation Channel (PCC) Family consists of several transporters ranging in size from 500 to over 4000 amino acyl residues (aas) in length and exhibiting between 5 and 18 transmembrane segments (TMSs). This family is a constituent of the Voltage-Gated Ion Channel (VIC) Superfamily. These transporters generally catalyze the export of cations. A representative list of proteins belonging to the PCC family can be found in the Transporter Classification Database.
Polycystic kidney disease 3 (autosomal dominant) is a protein that in humans is encoded by the PKD3 gene.

Shrawan Kumar, is an Indian-American geneticist, working in the fields of molecular and population genetics. He is known for his contributions in the discovery of two genes related to Branchio-oto-renal syndrome (BOR) and Autosomal Dominant Polycystic Kidney Disease (ADPKD2).