Kidney disease, or renal disease, technically referred to as nephropathy, is damage to or disease of a kidney. Nephritis is an inflammatory kidney disease and has several types according to the location of the inflammation. Inflammation can be diagnosed by blood tests. Nephrosis is non-inflammatory kidney disease. Nephritis and nephrosis can give rise to nephritic syndrome and nephrotic syndrome respectively. Kidney disease usually causes a loss of kidney function to some degree and can result in kidney failure, the complete loss of kidney function. Kidney failure is known as the end-stage of kidney disease, where dialysis or a kidney transplant is the only treatment option.

Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

A nephrectomy is the surgical removal of a kidney, performed to treat a number of kidney diseases including kidney cancer. It is also done to remove a normal healthy kidney from a living or deceased donor, which is part of a kidney transplant procedure.
Secondary hypertension is a type of hypertension which by definition is caused by an identifiable underlying primary cause. It is much less common than the other type, called essential hypertension, affecting only 5-10% of hypertensive patients. It has many different causes including endocrine diseases, kidney diseases, and tumors. It also can be a side effect of many medications.

Cystic kidney disease refers to a wide range of hereditary, developmental, and acquired conditions and with the inclusion of neoplasms with cystic changes, over 40 classifications and subtypes have been identified. Depending on the disease classification, the presentation may be at birth, or much later into adult life. Cystic disease may involve one or both kidneys and may, or may not, occur in the presence of other anomalies. A higher incidence is found in males and prevalence increases with age. Renal cysts have been reported in more than 50% of patients over the age of 50. Typically, cysts grow up to 2.88 mm annually and may cause related pain and/or hemorrhage.

Medullary cystic kidney disease (MCKD) is an autosomal dominant kidney disorder characterized by tubulointerstitial sclerosis leading to end-stage renal disease. Because the presence of cysts is neither an early nor a typical diagnostic feature of the disease, and because at least 4 different gene mutations may give rise to the condition, the name autosomal dominant tubulointerstitial kidney disease (ADTKD) has been proposed, to be appended with the underlying genetic variant for a particular individual. Importantly, if cysts are found in the medullary collecting ducts they can result in a shrunken kidney, unlike that of polycystic kidney disease. There are two known forms of medullary cystic kidney disease, mucin-1 kidney disease 1 (MKD1) and mucin-2 kidney disease/uromodulin kidney disease (MKD2). A third form of the disease occurs due to mutations in the gene encoding renin (ADTKD-REN), and has formerly been known as familial juvenile hyperuricemic nephropathy type 2.

Tolvaptan, sold under the brand name Samsca among others, is an aquaretic drug that functions as a selective, competitive vasopressin receptor 2 (V2) antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca. Tolvaptan, as Jynarque, was granted approval for medical use in the United States in April 2018.
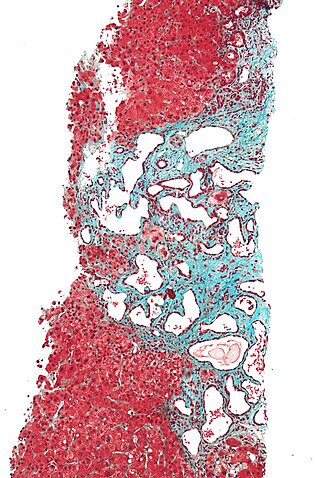
Polycystic liver disease (PLD) usually describes the presence of multiple cysts scattered throughout normal liver tissue. PLD is commonly seen in association with autosomal-dominant polycystic kidney disease, with a prevalence of 1 in 400 to 1000, and accounts for 8–10% of all cases of end-stage renal disease. The much rarer autosomal-dominant polycystic liver disease will progress without any kidney involvement.

Nephronophthisis is a genetic disorder of the kidneys which affects children. It is classified as a medullary cystic kidney disease. The disorder is inherited in an autosomal recessive fashion and, although rare, is the most common genetic cause of childhood kidney failure. It is a form of ciliopathy. Its incidence has been estimated to be 0.9 cases per million people in the United States, and 1 in 50,000 births in Canada.

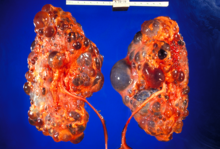
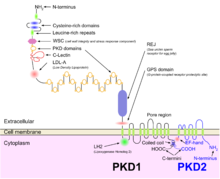
Polycystin 1 (PC1) is a protein that in humans is encoded by the PKD1 gene. Mutations of PKD1 are associated with most cases of autosomal dominant polycystic kidney disease, a severe hereditary disorder of the kidneys characterised by the development of renal cysts and severe kidney dysfunction.

Polycystin-2(PC2) is a protein that in humans is encoded by the PKD2 gene.

Polycystic kidney disease 2-like 1 protein also known as transient receptor potential polycystic 2 is a protein that in humans is encoded by the PKD2L1 gene.

Polycystic kidney disease is a genetic disorder in which the renal tubules become structurally abnormal, resulting in the development and growth of multiple cysts within the kidney. These cysts may begin to develop in utero, in infancy, in childhood, or in adulthood. Cysts are non-functioning tubules filled with fluid pumped into them, which range in size from microscopic to enormous, crushing adjacent normal tubules and eventually rendering them non-functional as well.
Autosomal recessive polycystic kidney disease (ARPKD) is the recessive form of polycystic kidney disease. It is associated with a group of congenital fibrocystic syndromes. Mutations in the PKHD1 cause ARPKD.

Lixivaptan (VPA-985) is an orally-active, non-peptide, selective vasopressin 2 receptor antagonist being developed as an investigational drug by Palladio Biosciences, Inc. (Palladio), a subsidiary of Centessa Pharmaceuticals plc. As of December 2021, lixivaptan is in Phase III clinical development for the treatment of Autosomal dominant polycystic kidney disease (ADPKD), the most common form of polycystic kidney disease. The U.S. Food and Drug Administration (FDA) has granted orphan drug designation to lixivaptan for the treatment of ADPKD.
Glomerulocystic kidney disease (GCKD) is a cystic disorder of the kidneys. GCKD involves cystic dilation of Bowman's capsule. It can occur with or without congenital abnormality.
The Polycystin Cation Channel (PCC) Family consists of several transporters ranging in size from 500 to over 4000 amino acyl residues (aas) in length and exhibiting between 5 and 18 transmembrane segments (TMSs). This family is a constituent of the Voltage-Gated Ion Channel (VIC) Superfamily. These transporters generally catalyze the export of cations. A representative list of proteins belonging to the PCC family can be found in the Transporter Classification Database.
Polycystic kidney disease 3 (autosomal dominant) is a protein that in humans is encoded by the PKD3 gene.

Thomas Martin Barratt was a British paediatrician and professor of paediatric nephrology. Barratt was most notable for developing a specialist service for children with kidney diseases in Britain, bringing peritoneal dialysis, haemodialysis, and later renal transplantation to ever younger children. Barratt was an early advocate for multidisciplinary care and developed a model that was later taken up by many other specialist centres across the world. His research led to a new treatments for many types of childhood kidney diseases., and for research into childhood Nephrotic syndrome and Hemolytic-uremic syndrome.

Shrawan Kumar, is an Indian-American geneticist, working in the fields of molecular and population genetics. He is known for his contributions in the discovery of two genes related to Branchio-oto-renal syndrome (BOR) and Autosomal Dominant Polycystic Kidney Disease (ADPKD2).