
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common, life-threatening inherited human disorders and the most common hereditary kidney disease. It is associated with large interfamilial and intrafamilial variability, which can be explained to a large extent by its genetic heterogeneity and modifier genes. It is also the most common of the inherited cystic kidney diseases — a group of disorders with related but distinct pathogenesis, characterized by the development of renal cysts and various extrarenal manifestations, which in case of ADPKD include cysts in other organs, such as the liver, seminal vesicles, pancreas, and arachnoid membrane, as well as other abnormalities, such as intracranial aneurysms and dolichoectasias, aortic root dilatation and aneurysms, mitral valve prolapse, and abdominal wall hernias. Over 50% of patients with ADPKD eventually develop end stage kidney disease and require dialysis or kidney transplantation. ADPKD is estimated to affect at least one in every 1000 individuals worldwide, making this disease the most common inherited kidney disorder with a diagnosed prevalence of 1:2000 and incidence of 1:3000-1:8000 in a global scale.

Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.
Chromosome 6 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 6 spans more than 172 million base pairs and represents between 5.5 and 6% of the total DNA in cells. It contains the major histocompatibility complex, which contains over 100 genes related to the immune response, and plays a vital role in organ transplantation.

Cystic kidney disease refers to a wide range of hereditary, developmental, and acquired conditions and with the inclusion of neoplasms with cystic changes, over 40 classifications and subtypes have been identified. Depending on the disease classification, the presentation may be at birth, or much later into adult life. Cystic disease may involve one or both kidneys and may, or may not, occur in the presence of other anomalies. A higher incidence is found in males and prevalence increases with age. Renal cysts have been reported in more than 50% of patients over the age of 50. Typically, cysts grow up to 2.88 mm annually and may cause related pain and/or hemorrhage.

Medullary cystic kidney disease (MCKD) is an autosomal dominant kidney disorder characterized by tubulointerstitial sclerosis leading to end-stage renal disease. Because the presence of cysts is neither an early nor a typical diagnostic feature of the disease, and because at least 4 different gene mutations may give rise to the condition, the name autosomal dominant tubulointerstitial kidney disease (ADTKD) has been proposed, to be appended with the underlying genetic variant for a particular individual. Importantly, if cysts are found in the medullary collecting ducts they can result in a shrunken kidney, unlike that of polycystic kidney disease. There are two known forms of medullary cystic kidney disease, mucin-1 kidney disease 1 (MKD1) and mucin-2 kidney disease/uromodulin kidney disease (MKD2). A third form of the disease occurs due to mutations in the gene encoding renin (ADTKD-REN), and has formerly been known as familial juvenile hyperuricemic nephropathy type 2.
Pendrin is an anion exchange protein that in humans is encoded by the SLC26A4 gene . Pendrin was initially identified as a sodium-independent chloride-iodide exchanger with subsequent studies showing that it also accepts formate and bicarbonate as substrates. Pendrin is similar to the Band 3 transport protein found in red blood cells. Pendrin is the protein which is mutated in Pendred syndrome, which is an autosomal recessive disorder characterized by sensorineural hearing loss, goiter and a partial organification problem detectable by a positive perchlorate test.
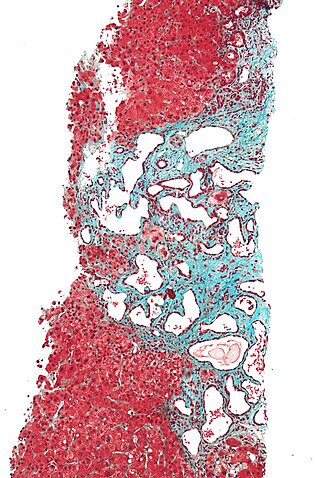
Polycystic liver disease (PLD) usually describes the presence of multiple cysts scattered throughout normal liver tissue. PLD is commonly seen in association with autosomal-dominant polycystic kidney disease, with a prevalence of 1 in 400 to 1000, and accounts for 8–10% of all cases of end-stage renal disease. The much rarer autosomal-dominant polycystic liver disease will progress without any kidney involvement.

Fibrocystin is a large, receptor-like protein that is thought to be involved in the tubulogenesis and/or maintenance of duct-lumen architecture of epithelium. FPC associates with the primary cilia of epithelial cells and co-localizes with the Pkd2 gene product polycystin-2 (PC2), suggesting that these two proteins may function in a common molecular pathway.

Dent's disease is a rare X-linked recessive inherited condition that affects the proximal renal tubules of the kidney. It is one cause of Fanconi syndrome, and is characterized by tubular proteinuria, excess calcium in the urine, formation of calcium kidney stones, nephrocalcinosis, and chronic kidney failure.
Alstrom syndrome 1 also known as ALMS1 is a protein which in humans is encoded by the ALMS1 gene.

Polycystin 1 (PC1) is a protein that in humans is encoded by the PKD1 gene. Mutations of PKD1 are associated with most cases of autosomal dominant polycystic kidney disease, a severe hereditary disorder of the kidneys characterised by the development of renal cysts and severe kidney dysfunction.

Polycystin-2(PC2) is a protein that in humans is encoded by the PKD2 gene.

Intraflagellar transport protein 88 homolog is a protein that is encoded by the IFT88 gene.

A ciliopathy is any genetic disorder that affects the cellular cilia or the cilia anchoring structures, the basal bodies, or ciliary function. Primary cilia are important in guiding the process of development, so abnormal ciliary function while an embryo is developing can lead to a set of malformations that can occur regardless of the particular genetic problem. The similarity of the clinical features of these developmental disorders means that they form a recognizable cluster of syndromes, loosely attributed to abnormal ciliary function and hence called ciliopathies. Regardless of the actual genetic cause, it is clustering of a set of characteristic physiological features which define whether a syndrome is a ciliopathy.
Polycystic kidney disease is a genetic disorder in which the renal tubules become structurally abnormal, resulting in the development and growth of multiple cysts within the kidney. These cysts may begin to develop in utero, in infancy, in childhood, or in adulthood. Cysts are non-functioning tubules filled with fluid pumped into them, which range in size from microscopic to enormous, crushing adjacent normal tubules and eventually rendering them non-functional as well.
A BBSome is a protein complex that operates in primary cilia biogenesis, homeostasis, and intraflagellar transport (IFT). The BBSome recognizes cargo proteins and signaling molecules like G-protein coupled receptors (GPCRs) on the ciliary membrane and helps transport them to and from the primary cilia. Primary cilia are nonmotile microtubule projections that function like antennae and are found in many types of cells. They receive various environmental signals to aid the cell in survival. They can detect photons by concentrating rhodopsin, a light receptor that converts photons into chemical signals, or odorants by concentrating olfactory receptors on the primary cilia surface. Primary cilia are also meaningful in cell development and signaling. They do not contain any way to make proteins within the primary cilia, so the BBSome aids in transporting essential proteins to, from, and within the cilia. Examples of cargo proteins that the BBSome is responsible for ferrying include smoothened, polycystic-1 (PC1), and several G-Protein coupled receptors (GPCRs) like somatostatin receptors (Sstr3), melanin-concentrating hormone receptor 1 (Mchr1), and neuropeptide Y2 receptor.
Glomerulocystic kidney disease (GCKD) is a cystic disorder of the kidneys. GCKD involves cystic dilation of Bowman's capsule. It can occur with or without congenital abnormality.
The Polycystin Cation Channel (PCC) Family consists of several transporters ranging in size from 500 to over 4000 amino acyl residues (aas) in length and exhibiting between 5 and 18 transmembrane segments (TMSs). This family is a constituent of the Voltage-Gated Ion Channel (VIC) Superfamily. These transporters generally catalyze the export of cations. A representative list of proteins belonging to the PCC family can be found in the Transporter Classification Database.
Polycystic kidney disease 3 (autosomal dominant) is a protein that in humans is encoded by the PKD3 gene.
HUPRA syndrome is a rare syndrome that was first described in 2010 in two infants of Palestinian origin from the same village in the Jerusalem area. One of the two infants' parents were related. It was later described in a third infant from the same village, whose parents were not related.


