
Motor neuron diseases or motor neurone diseases (MNDs) are a group of rare neurodegenerative disorders that selectively affect motor neurons, the cells which control voluntary muscles of the body. They include amyotrophic lateral sclerosis (ALS), progressive bulbar palsy (PBP), pseudobulbar palsy, progressive muscular atrophy (PMA), primary lateral sclerosis (PLS), spinal muscular atrophy (SMA) and monomelic amyotrophy (MMA), as well as some rarer variants resembling ALS.

Charcot–Marie–Tooth disease (CMT) is a hereditary motor and sensory neuropathy of the peripheral nervous system characterized by progressive loss of muscle tissue and touch sensation across various parts of the body. This disease is the most commonly inherited neurological disorder, affecting about one in 2,500 people. It is named after those who classically described it: the Frenchman Jean-Martin Charcot (1825–1893), his pupil Pierre Marie (1853–1940), and the Briton Howard Henry Tooth (1856–1925).
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder. The disease presents with progressive stiffness (spasticity) and contraction in the lower limbs. HSP is also known as hereditary spastic paraparesis, familial spastic paraplegia, French settlement disease, Strumpell disease, or Strumpell-Lorrain disease. The symptoms are a result of dysfunction of long axons in the spinal cord. The affected cells are the primary motor neurons; therefore, the disease is an upper motor neuron disease. HSP is not a form of cerebral palsy even though it physically may appear and behave much the same as spastic diplegia. The origin of HSP is different from cerebral palsy. Despite this, some of the same anti-spasticity medications used in spastic cerebral palsy are sometimes used to treat HSP symptoms.

Becker muscular dystrophy is an X-linked recessive inherited disorder characterized by slowly progressing muscle weakness of the legs and pelvis. It is a type of dystrophinopathy. This is caused by mutations in the dystrophin gene, which encodes the protein dystrophin. Becker muscular dystrophy is related to Duchenne muscular dystrophy in that both result from a mutation in the dystrophin gene, but has a milder course.
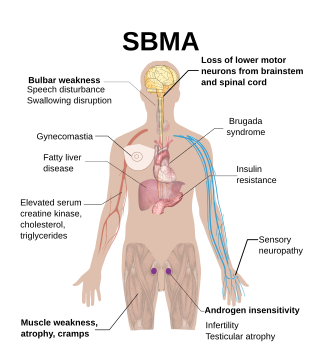
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.

Sandhoff disease is a lysosomal genetic, lipid storage disorder caused by the inherited deficiency to create functional beta-hexosaminidases A and B. These catabolic enzymes are needed to degrade the neuronal membrane components, ganglioside GM2, its derivative GA2, the glycolipid globoside in visceral tissues, and some oligosaccharides. Accumulation of these metabolites leads to a progressive destruction of the central nervous system and eventually to death. The rare autosomal recessive neurodegenerative disorder is clinically almost indistinguishable from Tay–Sachs disease, another genetic disorder that disrupts beta-hexosaminidases A and S. There are three subsets of Sandhoff disease based on when first symptoms appear: classic infantile, juvenile and adult late onset.
Myotonia congenita is a congenital neuromuscular channelopathy that affects skeletal muscles. It is a genetic disorder. The hallmark of the disease is the failure of initiated contraction to terminate, often referred to as delayed relaxation of the muscles (myotonia) and rigidity. Symptoms include delayed relaxation of the muscles after voluntary contraction (myotonia), and may also include stiffness, hypertrophy (enlargement), transient weakness in some forms of the disorder, severe masseter spasm, and cramping. The condition is sometimes referred to as fainting goat syndrome, as it is responsible for the eponymous 'fainting' seen in fainting goats when presented with a sudden stimulus. Of note, myotonia congenita has no association with malignant hyperthermia (MH).
Primary lateral sclerosis (PLS) is a very rare neuromuscular disease characterized by progressive muscle weakness in the voluntary muscles. PLS belongs to a group of disorders known as motor neuron diseases. Motor neuron diseases develop when the nerve cells that control voluntary muscle movement degenerate and die, causing weakness in the muscles they control.

Progressive muscular atrophy (PMA), also called Duchenne–Aran disease and Duchenne–Aran muscular atrophy, is a disorder characterised by the degeneration of lower motor neurons, resulting in generalised, progressive loss of muscle function.
Progressive bulbar palsy (PBP) is a medical condition. It belongs to a group of disorders known as motor neuron diseases. PBP is a disease that attacks the nerves supplying the bulbar muscles. These disorders are characterized by the degeneration of motor neurons in the cerebral cortex, spinal cord, brain stem, and pyramidal tracts. This specifically involves the glossopharyngeal nerve (IX), vagus nerve (X), and hypoglossal nerve (XII).
Pyruvate carboxylase deficiency is an inherited disorder that causes lactic acid to accumulate in the blood. High levels of these substances can damage the body's organs and tissues, particularly in the nervous system. Pyruvate carboxylase deficiency is a rare condition, with an estimated incidence of 1 in 250,000 births worldwide. Type A of the disease appears to be much more common in some Algonkian Indian tribes in eastern Canada, while the type B disease is more present in European populations.

Potassium-aggravated myotonia is a rare genetic disorder that affects skeletal muscle. Beginning in childhood or adolescence, people with this condition experience bouts of sustained muscle tensing (myotonia) that prevent muscles from relaxing normally. Myotonia causes muscle stiffness, often painful, that worsens after exercise and may be aggravated by eating potassium-rich foods such as bananas and potatoes. Stiffness occurs in skeletal muscles throughout the body. Potassium-aggravated myotonia ranges in severity from mild episodes of muscle stiffness to severe, disabling disease with frequent attacks. Potassium-aggravated myotonia may, in some cases, also cause paradoxical myotonia, in which myotonia becomes more severe at the time of movement instead of after movement has ceased. Unlike some other forms of myotonia, potassium-aggravated myotonia is not associated with episodes of muscle weakness.
Hereditary sensory and autonomic neuropathy (HSAN) or hereditary sensory neuropathy (HSN) is a condition used to describe any of the types of this disease which inhibit sensation.
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease (MND) or Lou Gehrig's disease, is a neurodegenerative disease that results in the progressive loss of motor neurons that control voluntary muscles. ALS is the most common form of the motor neuron diseases. Early symptoms of ALS include stiff muscles, muscle twitches, and gradual increasing weakness and muscle wasting. Limb-onset ALS begins with weakness in the arms or legs, while bulbar-onset ALS begins with difficulty speaking or swallowing. Around half of people with ALS develop at least mild difficulties with thinking and behavior, and about 15% develop frontotemporal dementia. Motor neuron loss continues until the ability to eat, speak, move, and finally the ability to breathe is lost with the cause of early death usually being respiratory failure.

C9orf72 is a protein which in humans is encoded by the gene C9orf72.
Multisystem proteinopathy (MSP) is a dominantly inherited, pleiotropic, degenerative disorder of humans that can affect muscle, bone, and/or the central nervous system. MSP can manifest clinically as classical amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), inclusion body myopathy (IBM), Paget's disease of bone (PDB), or as a combination of these disorders. Historically, several different names have been used to describe MSP, most commonly "inclusion body myopathy with early-onset Paget disease and frontotemporal dementia (IBMPFD)" or "inclusion body myopathy with frontotemporal dementia, Paget's disease of bone, and amyotrophic lateral sclerosis (IBMPFD/ALS)." However, IBMPFD and IBMPFD/ALS are now considered outdated classifications and are more properly referred to as MSP, as the disease is clinically heterogeneous and its phenotypic spectrum extends beyond IBM, PDB, FTD, and ALS to include motor neuron disease, Parkinson's disease features, and ataxia features. Although MSP is rare, growing interest in this syndrome derives from the molecular insights the condition provides into the etiological relationship between common age-related degenerative diseases of muscle, bone, and brain.

Neurodegenerative diseases are a heterogeneous group of complex disorders linked by the degeneration of neurons in either the peripheral nervous system or the central nervous system. Their underlying causes are extremely variable and complicated by various genetic and/or environmental factors. These diseases cause progressive deterioration of the neuron resulting in decreased signal transduction and in some cases even neuronal death. Peripheral nervous system diseases may be further categorized by the type of nerve cell affected by the disorder. Effective treatment of these diseases is often prevented by lack of understanding of the underlying molecular and genetic pathology. Epigenetic therapy is being investigated as a method of correcting the expression levels of misregulated genes in neurodegenerative diseases.
Adult polyglucosan body disease (APBD) is a rare genetic glycogen storage disorder caused by an inborn error of metabolism. Symptoms can emerge any time after the age of 30; early symptoms include trouble controlling urination, trouble walking, and lack of sensation in the legs. People eventually develop dementia.
There are more than 25 genes known to be associated with amyotrophic lateral sclerosis (ALS) as of June 2018, which collectively account for about 70% of cases of familial ALS (fALS) and 10% of cases of sporadic ALS (sALS). About 5–10% of cases of ALS are directly inherited. Overall, first-degree relatives of an individual with ALS have a 1% risk of developing ALS. ALS has an oligogenic mode of inheritance, meaning that mutations in two or more genes are required to cause disease.
Research on amyotrophic lateral sclerosis (ALS) has focused on animal models of the disease, its mechanisms, ways to diagnose and track it, and treatments.
