
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Sabinas brittle hair syndrome, also called Sabinas syndrome or brittle hair-mental deficit syndrome, is an autosomal recessive congenital disorder affecting the integumentary system.

Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

Menkes disease (MNK), also known as Menkes syndrome, is an X-linked recessive disorder caused by mutations in genes coding for the copper-transport protein ATP7A, leading to copper deficiency. Characteristic findings include kinky hair, growth failure, and nervous system deterioration. Like all X-linked recessive conditions, Menkes disease is more common in males than in females. The disorder was first described by John Hans Menkes in 1962.

Isovaleric acidemia is a rare autosomal recessive metabolic disorder which disrupts or prevents normal metabolism of the branched-chain amino acid leucine. It is a classical type of organic acidemia.

Maple syrup urine disease (MSUD) is an autosomal recessive metabolic disorder affecting branched-chain amino acids. It is one type of organic acidemia. The condition gets its name from the distinctive sweet odor of affected infants' urine and earwax, particularly prior to diagnosis and during times of acute illness. It was described by John Menkes in the 1950s.

Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine. It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.

Hartnup disease is an autosomal recessive metabolic disorder affecting the absorption of nonpolar amino acids. Niacin is a precursor to nicotinamide, a necessary component of NAD+.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.
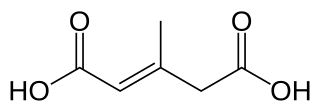
3-Methylglutaconic aciduria (MGA) is any of at least five metabolic disorders that impair the body's ability to make energy in the mitochondria. As a result of this impairment, 3-methylglutaconic acid and 3-methylglutaric acid build up and can be detected in the urine.

Glycine encephalopathy is a rare autosomal recessive disorder of glycine metabolism. After phenylketonuria, glycine encephalopathy is the second most common disorder of amino acid metabolism. The disease is caused by defects in the glycine cleavage system, an enzyme responsible for glycine catabolism. There are several forms of the disease, with varying severity of symptoms and time of onset. The symptoms are exclusively neurological in nature, and clinically this disorder is characterized by abnormally high levels of the amino acid glycine in bodily fluids and tissues, especially the cerebrospinal fluid.

SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. These characteristics are what make up the acronym SCARF. It shares some features with Lenz-Majewski hyperostotic dwarfism. It is a very rare disease with an incidence rate of approximately one in a million newborns. It has been clinically described in two males who were maternal cousins, as well as a 3-month-old female. Babies affected by this syndrome tend to have very loose skin, giving them an elderly facial appearance. Possible complications include dyspnea, abdominal hernia, heart disorders, joint disorders, and dislocations of multiple joints. It is believed that this disease's inheritance is X-linked recessive.
Papillon–Lefèvre syndrome (PLS), also known as palmoplantar keratoderma with periodontitis, is an autosomal recessive genetic disorder caused by a deficiency in cathepsin C.

Dent's disease is a rare X-linked recessive inherited condition that affects the proximal renal tubules of the kidney. It is one cause of Fanconi syndrome, and is characterized by tubular proteinuria, excess calcium in the urine, formation of calcium kidney stones, nephrocalcinosis, and chronic kidney failure.
Hypertryptophanemia is a rare autosomal recessive metabolic disorder that results in a massive buildup of the amino acid tryptophan in the blood, with associated symptoms and tryptophanuria.
Peeling skin syndrome is an autosomal recessive disorder characterized by lifelong peeling of the stratum corneum, and may be associated with pruritus, short stature, and easily removed anagen hair.

Aminolevulinic acid dehydratase deficiency porphyria is an extremely rare autosomal recessive metabolic disorder that results from inappropriately low levels of the enzyme delta-aminolevulinic acid dehydratase (ALAD), which is required for normal heme synthesis. This deficiency results in the accumulation of a toxic metabolic precursor in the heme synthesis pathway called aminolevulinic acid (ALA). Lead poisoning can also disrupt ALAD and result in elevated ALA causing the same symptoms. Heme is a component of hemoglobin which carries oxygen in red blood cells.

Monocarboxylate transporter 10, also known as aromatic amino acid transporter 1 and T-type amino acid transporter 1 (TAT1) and solute carrier family 16 member 10 (SLC16A10), is a protein that in humans is encoded by the SLC16A10 gene. SLC16A10 is a member of the solute carrier family.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is disease of the arteries in the brain, which causes tissue loss in the subcortical region of the brain and the destruction of myelin in the CNS. CARASIL is characterized by symptoms such as gait disturbances, hair loss, low back pain, dementia, and stroke. CARASIL is a rare disease, having only been diagnosed in about 50 patients, of which ten have been genetically confirmed. Most cases have been reported in Japan, but Chinese and caucasian individuals have also been diagnosed with the disease. CARASIL is inherited in an autosomal recessive pattern. There is currently no cure for CARASIL. Other names for CARASIL include familial young-adult-onset arteriosclerotic leukoencephalopathy with alopecia and lumbago without arterial hypertension, Nemoto disease and Maeda syndrome.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
