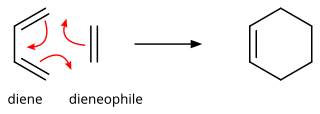
In organic chemistry, the Diels–Alder reaction is a chemical reaction between a conjugated diene and a substituted alkene, commonly termed the dienophile, to form a substituted cyclohexene derivative. It is the prototypical example of a pericyclic reaction with a concerted mechanism. More specifically, it is classified as a thermally-allowed [4+2] cycloaddition with Woodward–Hoffmann symbol [π4s + π2s]. It was first described by Otto Diels and Kurt Alder in 1928. For the discovery of this reaction, they were awarded the Nobel Prize in Chemistry in 1950. Through the simultaneous construction of two new carbon–carbon bonds, the Diels–Alder reaction provides a reliable way to form six-membered rings with good control over the regio- and stereochemical outcomes. Consequently, it has served as a powerful and widely applied tool for the introduction of chemical complexity in the synthesis of natural products and new materials. The underlying concept has also been applied to π-systems involving heteroatoms, such as carbonyls and imines, which furnish the corresponding heterocycles; this variant is known as the hetero-Diels–Alder reaction. The reaction has also been generalized to other ring sizes, although none of these generalizations have matched the formation of six-membered rings in terms of scope or versatility. Because of the negative values of ΔH° and ΔS° for a typical Diels–Alder reaction, the microscopic reverse of a Diels–Alder reaction becomes favorable at high temperatures, although this is of synthetic importance for only a limited range of Diels-Alder adducts, generally with some special structural features; this reverse reaction is known as the retro-Diels–Alder reaction.

In theoretical chemistry, a conjugated system is a system of connected p-orbitals with delocalized electrons in a molecule, which in general lowers the overall energy of the molecule and increases stability. It is conventionally represented as having alternating single and multiple bonds. Lone pairs, radicals or carbenium ions may be part of the system, which may be cyclic, acyclic, linear or mixed. The term "conjugated" was coined in 1899 by the German chemist Johannes Thiele.
In organic chemistry, a cycloaddition is a chemical reaction in which "two or more unsaturated molecules combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity". The resulting reaction is a cyclization reaction. Many but not all cycloadditions are concerted and thus pericyclic. Nonconcerted cycloadditions are not pericyclic. As a class of addition reaction, cycloadditions permit carbon–carbon bond formation without the use of a nucleophile or electrophile.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.
In chemistry, frontier molecular orbital theory is an application of molecular orbital theory describing HOMO–LUMO interactions.

Germylenes are a class of germanium(II) compounds with the general formula :GeR2. They are heavier carbene analogs. However, unlike carbenes, whose ground state can be either singlet or triplet depending on the substituents, germylenes have exclusively a singlet ground state. Unprotected carbene analogs, including germylenes, has a dimerization nature. Free germylenes can be isolated under the stabilization of steric hindrance or electron donation. The synthesis of first stable free dialkyl germylene was reported by Jutzi, et al in 1991.
The inverse electron demand Diels–Alder reaction, or DAINV or IEDDA is an organic chemical reaction, in which two new chemical bonds and a six-membered ring are formed. It is related to the Diels–Alder reaction, but unlike the Diels–Alder reaction, the DAINV is a cycloaddition between an electron-rich dienophile and an electron-poor diene. During a DAINV reaction, three pi-bonds are broken, and two sigma bonds and one new pi-bond are formed. A prototypical DAINV reaction is shown on the right.
A metal-centered cycloaddition is a subtype of the more general class of cycloaddition reactions. In such reactions "two or more unsaturated molecules unite directly to form a ring", incorporating a metal bonded to one or more of the molecules. Cycloadditions involving metal centers are a staple of organic and organometallic chemistry, and are involved in many industrially-valuable synthetic processes.
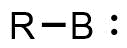
A borylene is the boron analogue of a carbene. The general structure is R-B: with R an organic moiety and B a boron atom with two unshared electrons. Borylenes are of academic interest in organoboron chemistry. A singlet ground state is predominant with boron having two vacant sp2 orbitals and one doubly occupied one. With just one additional substituent the boron is more electron deficient than the carbon atom in a carbene. For this reason stable borylenes are more uncommon than stable carbenes. Some borylenes such as boron monofluoride (BF) and boron monohydride (BH) the parent compound also known simply as borylene, have been detected in microwave spectroscopy and may exist in stars. Other borylenes exist as reactive intermediates and can only be inferred by chemical trapping.
Digermynes are a class of compounds that are regarded as the heavier digermanium analogues of alkynes. The parent member of this entire class is HGeGeH, which has only been characterized computationally, but has revealed key features of the whole class. Because of the large interatomic repulsion between two Ge atoms, only kinetically stabilized digermyne molecules can be synthesized and characterized by utilizing bulky protecting groups and appropriate synthetic methods, for example, reductive coupling of germanium(II) halides.
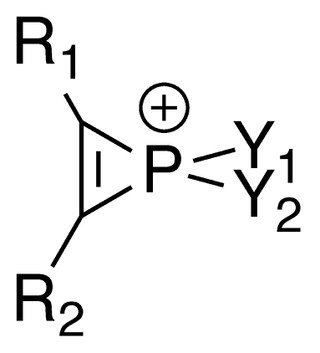
Phosphirenium ions are a series of organophosphorus compounds containing unsaturated three-membered ring phosphorus (V) heterocycles and σ*-aromaticity is believed to be present in such molecules. Many of the salts containing phosphirenium ions have been isolated and characterized by NMR spectroscopy and X-ray crystallography.

Phosphasilenes or silylidenephosphanes are a class of compounds with silicon-phosphorus double bonds. Since the electronegativity of phosphorus (2.1) is higher than that of silicon (1.9), the "Si=P" moiety of phosphasilene is polarized. The degree of polarization can be tuned by altering the coordination numbers of the Si and P centers, or by modifying the electronic properties of the substituents. The phosphasilene Si=P double bond is highly reactive, yet with the choice of proper substituents, it can be stabilized via donor-acceptor interaction or by steric congestion.

Tellurophenes are the tellurium analogue of thiophenes and selenophenes.

An N-Heterocyclic silylene (NHSi) is an uncharged heterocyclic chemical compound consisting of a divalent silicon atom bonded to two nitrogen atoms. The isolation of the first stable NHSi, also the first stable dicoordinate silicon compound, was reported in 1994 by Michael Denk and Robert West three years after Anthony Arduengo first isolated an N-heterocyclic carbene, the lighter congener of NHSis. Since their first isolation, NHSis have been synthesized and studied with both saturated and unsaturated central rings ranging in size from 4 to 6 atoms. The stability of NHSis, especially 6π aromatic unsaturated five-membered examples, make them useful systems to study the structure and reactivity of silylenes and low-valent main group elements in general. Though not used outside of academic settings, complexes containing NHSis are known to be competent catalysts for industrially important reactions. This article focuses on the properties and reactivity of five-membered NHSis.

The triboracyclopropenyl fragment is a cyclic structural motif in boron chemistry, named for its geometric similarity to cyclopropene. In contrast to nonplanar borane clusters that exhibit higher coordination numbers at boron (e.g., through 3-center 2-electron bonds to bridging hydrides or cations), triboracyclopropenyl-type structures are rings of three boron atoms where substituents at each boron are also coplanar to the ring. Triboracyclopropenyl-containing compounds are extreme cases of inorganic aromaticity. They are the lightest and smallest cyclic structures known to display the bonding and magnetic properties that originate from fully delocalized electrons in orbitals of σ and π symmetry. Although three-membered rings of boron are frequently so highly strained as to be experimentally inaccessible, academic interest in their distinctive aromaticity and possible role as intermediates of borane pyrolysis motivated extensive computational studies by theoretical chemists. Beginning in the late 1980s with mass spectrometry work by Anderson et al. on all-boron clusters, experimental studies of triboracyclopropenyls were for decades exclusively limited to gas-phase investigations of the simplest rings (ions of B3). However, more recent work has stabilized the triboracyclopropenyl moiety via coordination to donor ligands or transition metals, dramatically expanding the scope of its chemistry.
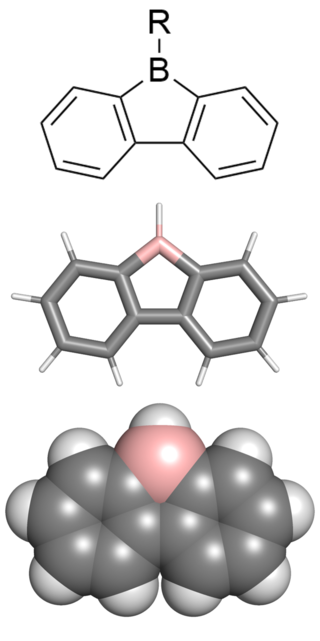
9-borafluorenes are a class of boron-containing heterocycles consisting of a tricyclic system with a central BC4 ring with two fused arene groups. 9-borafluorenes can be thought of as a borole with two fused arene rings, or as a trigonal planar boron atom with an empty p orbital bridging two biphenyl rings. However, 9-borafluorenes are generally less reactive than boroles due to less antiaromatic character and Lewis acidity. Containing highly conjugated π systems, 9-borafluorenes possess interesting photophysical properties. In addition, 9-borafluorenes are good Lewis acids. This combination of properties enables potential uses such as in light-emitting materials, solar cells, and sensors for some molecules.

Superelectrophilic anions are a class of molecular ions that exhibit highly electrophilic reaction behavior despite their overall negative charge. Thus, they are even able to bind the unreactive noble gases or molecular nitrogen at room temperature. The only representatives known so far are the fragment ions of the type [B12X11]– derived from the closo-dodecaborate dianions [B12X12]2–. X represents a substituent connected to a boron atom (cf. Fig. 1). For this reason, the following article deals exclusively with superelectrophilic anions of this type.

Boraacenes are polycyclic aromatic hydrocarbons containing at least one boron atom. Structurally, they are related to acenes, linearly fused benzene rings. However, the boron atom is electron deficient and may act as a Lewis Acid when compared to carbon. This results in slightly less negative charge within the ring, smaller HOMO-LUMO gaps, as well as differences in redox chemistry when compared to their acene analogues. When incorporated into acenes, Boron maintains the planarity and aromaticity of carbon acenes, while adding an empty p-orbital, which can be utilized for the fine tuning of organic semiconductor band gaps. Due to this empty p orbital, however, it is also highly reactive when exposed to nucleophiles like water or normal atmosphere, as it will readily be attacked by oxygen, which must be addressed to maintain its stability.

1,3-Diphospha-2,4-diboretanes, or B2P2, is a class of 4-member cyclic compounds of alternating boron and phosphorus atoms. They are often found as dimers during the synthesis of boraphosphenes (RB=PR'). Compounds can exhibit localized singlet diradical character (diradicaloid) between the boron atoms in the solution and solid state.

Aluminylenes are a sub-class of aluminium(I) compounds that feature singly-coordinated aluminium atoms with a lone pair of electrons. As aluminylenes exhibit two unoccupied orbitals, they are not strictly aluminium analogues of carbenes until stabilized by a Lewis base to form aluminium(I) nucleophiles. The lone pair and two empty orbitals on the aluminium allow for ambiphilic bonding where the aluminylene can act as both an electrophile and a nucleophile. Aluminylenes have also been reported under the names alumylenes and alanediyl.






![Resonance forms in the Borole dianion. Contributions to overall structure are 30.30%, 30.71% and 13.04% respectively going from left to right as determed using NRT. Analyses were performed using NBO7 on a [C4BH5] structure optimised using BP86-D3BJ and def2- TZVPP basis set. Resonance in borole 2-.png](http://upload.wikimedia.org/wikipedia/commons/thumb/6/66/Resonance_in_borole_2-.png/464px-Resonance_in_borole_2-.png)










