
Hydrogenation is a chemical reaction between molecular hydrogen (H2) and another compound or element, usually in the presence of a catalyst such as nickel, palladium or platinum. The process is commonly employed to reduce or saturate organic compounds. Hydrogenation typically constitutes the addition of pairs of hydrogen atoms to a molecule, often an alkene. Catalysts are required for the reaction to be usable; non-catalytic hydrogenation takes place only at very high temperatures. Hydrogenation reduces double and triple bonds in hydrocarbons.

Wilkinson's catalyst (chloridotris(triphenylphosphene)rhodium(I)) is a coordination complex of rhodium with the formula [RhCl(PPh3)3], where 'Ph' denotes a phenyl group. It is a red-brown colored solid that is soluble in hydrocarbon solvents such as benzene, and more so in tetrahydrofuran or chlorinated solvents such as dichloromethane. The compound is widely used as a catalyst for hydrogenation of alkenes. It is named after chemist and Nobel laureate Sir Geoffrey Wilkinson, who first popularized its use.
In organic chemistry, hydroboration refers to the addition of a hydrogen-boron bond to certain double and triple bonds involving carbon. This chemical reaction is useful in the organic synthesis of organic compounds.
In chemistry, transfer hydrogenation is a chemical reaction involving the addition of hydrogen to a compound from a source other than molecular H2. It is applied in laboratory and industrial organic synthesis to saturate organic compounds and reduce ketones to alcohols, and imines to amines. It avoids the need for high-pressure molecular H2 used in conventional hydrogenation. Transfer hydrogenation usually occurs at mild temperature and pressure conditions using organic or organometallic catalysts, many of which are chiral, allowing efficient asymmetric synthesis. It uses hydrogen donor compounds such as formic acid, isopropanol or dihydroanthracene, dehydrogenating them to CO2, acetone, or anthracene respectively. Often, the donor molecules also function as solvents for the reaction. A large scale application of transfer hydrogenation is coal liquefaction using "donor solvents" such as tetralin.

Tris(pentafluorophenyl)borane, sometimes referred to as "BCF", is the chemical compound (C6F5)3B. It is a white, volatile solid. The molecule consists of three pentafluorophenyl groups attached in a "paddle-wheel" manner to a central boron atom; the BC3 core is planar. It has been described as the “ideal Lewis acid” because of its high thermal stability and the relative inertness of the B-C bonds. Related fluoro-substituted boron compounds, such as those containing B−CF3 groups, decompose with formation of B-F bonds. Tris(pentafluorophenyl)borane is thermally stable at temperatures well over 200 °C, resistant to oxygen, and water-tolerant.
Organophosphines are organophosphorus compounds with the formula PRnH3−n, where R is an organic substituent. These compounds can be classified according to the value of n: primary phosphines (n = 1), secondary phosphines (n = 2), tertiary phosphines (n = 3). All adopt pyramidal structures. Organophosphines are generally colorless, lipophilic liquids or solids. The parent of the organophosphines is phosphine (PH3).
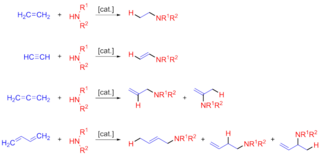
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.
Boroles represent a class of molecules known as metalloles, which are heterocyclic 5-membered rings. As such, they can be viewed as structural analogs of cyclopentadiene, pyrrole or furan, with boron replacing a carbon, nitrogen and oxygen atom respectively. They are isoelectronic with the cyclopentadienyl cation C5H+5(Cp+) and comprise four π electrons. Although Hückel's rule cannot be strictly applied to borole, it is considered to be antiaromatic due to having 4 π electrons. As a result, boroles exhibit unique electronic properties not found in other metalloles.

The Shvo catalyst is an organoruthenium compound that catalyzes the hydrogenation of polar functional groups including aldehydes, ketones and imines. The compound is of academic interest as an early example of a catalyst for transfer hydrogenation that operates by an "outer sphere mechanism". Related derivatives are known where p-tolyl replaces some of the phenyl groups. Shvo's catalyst represents a subset of homogeneous hydrogenation catalysts that involves both metal and ligand in its mechanism.

In chemistry, the hydrogenation of carbon–nitrogen double bonds is the addition of the elements of dihydrogen (H2) across a carbon–nitrogen double bond, forming amines or amine derivatives. Although a variety of general methods have been developed for the enantioselective hydrogenation of ketones, methods for the hydrogenation of carbon–nitrogen double bonds are less general. Hydrogenation of imines is complicated by both syn/anti isomerization and tautomerization to enamines, which may be hydrogenated with low enantioselectivity in the presence of a chiral catalyst. Additionally, the substituent attached to nitrogen affects both the reactivity and spatial properties of the imine, complicating the development of a general catalyst system for imine hydrogenation. Despite these challenges, methods have been developed that address particular substrate classes, such as N-aryl, N-alkyl, and endocyclic imines.
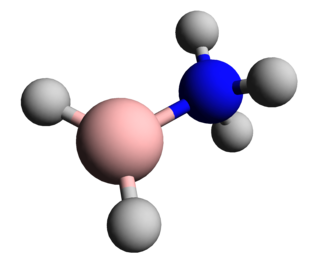
In chemistry, a boranylium ion is an inorganic cation with the chemical formula BR+
2, where R represents a non-specific substituent. Being electron-deficient, boranylium ions form adducts with Lewis bases. Boranylium ions have historical names that depend on the number of coordinated ligands:
Dehydrogenation of amine-boranes or dehydrocoupling of amine-boranes is a chemical process in main group and organometallic chemistry wherein dihydrogen is released by the coupling of two or more amine-borane adducts. This process is of due to the potential of using amine-boranes for hydrogen storage.
In chemistry, the Gutmann–Beckett method is an experimental procedure used by chemists to assess the Lewis acidity of molecular species. Triethylphosphine oxide is used as a probe molecule and systems are evaluated by 31P-NMR spectroscopy. In 1975, Viktor Gutmann used 31P-NMR spectroscopy to parameterize Lewis acidity of solvents by acceptor numbers (AN). In 1996, Michael A. Beckett recognised its more generally utility and adapted the procedure so that it could be easily applied to molecular species, when dissolved in weakly Lewis acidic solvents. The term Gutmann–Beckett method was first used in chemical literature in 2007.
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.
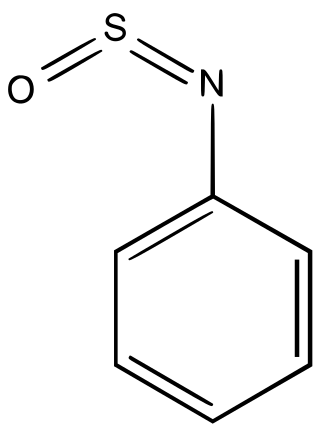
Sulfinylamines are organosulfur compounds with the formula RNSO where R = an organic substituent. These compounds are, formally speaking, derivatives of HN=S=O, i.e. analogues of sulfur dioxide and of sulfur diimide. A common example is N-sulfinylaniline. Sulfinyl amines are dienophile. They undergo [2+2] cycloaddition to ketenes.
Germanium(II) hydrides, also called germylene hydrides, are a class of Group 14 compounds consisting of low-valent germanium and a terminal hydride. They are also typically stabilized by an electron donor-acceptor interaction between the germanium atom and a large, bulky ligand.
Metal-ligand cooperativity (MLC) is a mode of reactivity in which a metal and ligand of a complex are both involved in the bond breaking or bond formation of a substrate during the course of a reaction. This ligand is an actor ligand rather than a spectator, and the reaction is generally only deemed to contain MLC if the actor ligand is doing more than leaving to provide an open coordination site. MLC is also referred to as "metal-ligand bifunctional catalysis." Note that MLC is not to be confused with cooperative binding.
Heteroatomic multiple bonding between group 13 and group 15 elements are of great interest in synthetic chemistry due to their isoelectronicity with C-C multiple bonds. Nevertheless, the difference of electronegativity between group 13 and 15 leads to different character of bondings comparing to C-C multiple bonds. Because of the ineffective overlap between p𝝅 orbitals and the inherent lewis acidity/basicity of group 13/15 elements, the synthesis of compounds containing such multiple bonds is challenging and subject to oligomerization. The most common example of compounds with 13/15 group multiple bonds are those with B=N units. The boron-nitrogen-hydride compounds are candidates for hydrogen storage. In contrast, multiple bonding between aluminium and nitrogen Al=N, Gallium and nitrogen (Ga=N), boron and phosphorus (B=P), or boron and arsenic (B=As) are less common.
In organic chemistry, carboboration describes an addition of both a carbon and a boron moiety to certain carbon-containing double and triple bonds, such as alkenes, alkynes, and allenes.

An N-heterocyclic olefin (NHO) is a neutral heterocyclic compound with a highly polarized, electron-rich C=C olefin attached to a heterocycle made up of two nitrogen atoms. A derivative of N-heterocyclic carbenes (NHCs), NHO was first synthesized in 1961 by Horst Böhme and Fritz Soldan, but the term NHO was not used until 2011 by Eric Rivard and coworkers. Since its discovery, NHOs have been applied in organocatalysis, metal ligation, and polymerization.










