
In organic chemistry, an alkyne is an unsaturated hydrocarbon containing at least one carbon—carbon triple bond. The simplest acyclic alkynes with only one triple bond and no other functional groups form a homologous series with the general chemical formula CnH2n−2. Alkynes are traditionally known as acetylenes, although the name acetylene also refers specifically to C2H2, known formally as ethyne using IUPAC nomenclature. Like other hydrocarbons, alkynes are generally hydrophobic.
Hydroboration–oxidation reaction is a two-step hydration reaction that converts an alkene into an alcohol. The process results in the syn addition of a hydrogen and a hydroxyl group where the double bond had been. Hydroboration–oxidation is an anti-Markovnikov reaction, with the hydroxyl group attaching to the less-substituted carbon. The reaction thus provides a more stereospecific and complementary regiochemical alternative to other hydration reactions such as acid-catalyzed addition and the oxymercuration–reduction process. The reaction was first reported by Herbert C. Brown in the late 1950s and it was recognized in his receiving the Nobel Prize in Chemistry in 1979.
The 1,3-dipolar cycloaddition is a chemical reaction between a 1,3-dipole and a dipolarophile to form a five-membered ring. The earliest 1,3-dipolar cycloadditions were described in the late 19th century to the early 20th century, following the discovery of 1,3-dipoles. Mechanistic investigation and synthetic application were established in the 1960s, primarily through the work of Rolf Huisgen. Hence, the reaction is sometimes referred to as the Huisgen cycloaddition. 1,3-dipolar cycloaddition is an important route to the regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives. The dipolarophile is typically an alkene or alkyne, but can be other pi systems. When the dipolarophile is an alkyne, aromatic rings are generally produced.

Organoboron chemistry or organoborane chemistry is the chemistry of organoboron compounds or organoboranes, which are chemical compounds of boron and carbon that are organic derivatives of borane (BH3), for example trialkyl boranes..
In organic chemistry, hydroboration refers to the addition of a hydrogen-boron bond to certain double and triple bonds involving carbon. This chemical reaction is useful in the organic synthesis of organic compounds.

The Corey–Itsuno reduction, also known as the Corey–Bakshi–Shibata (CBS) reduction, is a chemical reaction in which a prochiral ketone is enantioselectively reduced to produce the corresponding chiral, non-racemic alcohol. The oxazaborolidine reagent which mediates the enantioselective reduction of ketones was previously developed by the laboratory of Itsuno and thus this transformation may more properly be called the Itsuno-Corey oxazaborolidine reduction.

Azomethine ylides are nitrogen-based 1,3-dipoles, consisting of an iminium ion next to a carbanion. They are used in 1,3-dipolar cycloaddition reactions to form five-membered heterocycles, including pyrrolidines and pyrrolines. These reactions are highly stereo- and regioselective, and have the potential to form four new contiguous stereocenters. Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals. Azomethine ylides can be generated from many sources, including aziridines, imines, and iminiums. They are often generated in situ, and immediately reacted with dipolarophiles.
A carbometallation is any reaction where a carbon-metal bond reacts with a carbon-carbon π-bond to produce a new carbon-carbon σ-bond and a carbon-metal σ-bond. The resulting carbon-metal bond can undergo further carbometallation reactions or it can be reacted with a variety of electrophiles including halogenating reagents, carbonyls, oxygen, and inorganic salts to produce different organometallic reagents. Carbometallations can be performed on alkynes and alkenes to form products with high geometric purity or enantioselectivity, respectively. Some metals prefer to give the anti-addition product with high selectivity and some yield the syn-addition product. The outcome of syn and anti- addition products is determined by the mechanism of the carbometallation.
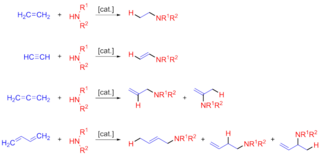
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.
A frustrated Lewis pair (FLP) is a compound or mixture containing a Lewis acid and a Lewis base that, because of steric hindrance, cannot combine to form a classical adduct. Many kinds of FLPs have been devised, and many simple substrates exhibit activation.

Organocobalt chemistry is the chemistry of organometallic compounds containing a carbon to cobalt chemical bond. Organocobalt compounds are involved in several organic reactions and the important biomolecule vitamin B12 has a cobalt-carbon bond. Many organocobalt compounds exhibit useful catalytic properties, the preeminent example being dicobalt octacarbonyl.
Organogold chemistry is the study of compounds containing gold–carbon bonds. They are studied in academic research, but have not received widespread use otherwise. The dominant oxidation states for organogold compounds are I with coordination number 2 and a linear molecular geometry and III with CN = 4 and a square planar molecular geometry.
Metal carbon dioxide complexes are coordination complexes that contain carbon dioxide ligands. Aside from the fundamental interest in the coordination chemistry of simple molecules, studies in this field are motivated by the possibility that transition metals might catalyze useful transformations of CO2. This research is relevant both to organic synthesis and to the production of "solar fuels" that would avoid the use of petroleum-based fuels.
In chemistry, metal-catalysed hydroboration is a reaction used in organic synthesis. It is one of several examples of homogeneous catalysis.
Metal-catalyzed C–H borylation reactions are transition metal catalyzed organic reactions that produce an organoboron compound through functionalization of aliphatic and aromatic C–H bonds and are therefore useful reactions for carbon–hydrogen bond activation. Metal-catalyzed C–H borylation reactions utilize transition metals to directly convert a C–H bond into a C–B bond. This route can be advantageous compared to traditional borylation reactions by making use of cheap and abundant hydrocarbon starting material, limiting prefunctionalized organic compounds, reducing toxic byproducts, and streamlining the synthesis of biologically important molecules. Boronic acids, and boronic esters are common boryl groups incorporated into organic molecules through borylation reactions. Boronic acids are trivalent boron-containing organic compounds that possess one alkyl substituent and two hydroxyl groups. Similarly, boronic esters possess one alkyl substituent and two ester groups. Boronic acids and esters are classified depending on the type of carbon group (R) directly bonded to boron, for example alkyl-, alkenyl-, alkynyl-, and aryl-boronic esters. The most common type of starting materials that incorporate boronic esters into organic compounds for transition metal catalyzed borylation reactions have the general formula (RO)2B-B(OR)2. For example, bis(pinacolato)diboron (B2Pin2), and bis(catecholato)diborane (B2Cat2) are common boron sources of this general formula.
Hydrophosphination is the insertion of a carbon-carbon multiple bond into a phosphorus-hydrogen bond forming a new phosphorus-carbon bond. Like other hydrofunctionalizations, the rate and regiochemistry of the insertion reaction is influenced by the catalyst. Catalysts take many forms, but most prevalent are bases and free-radical initiators. Most hydrophosphinations involve reactions of phosphine (PH3).

In organometallic chemistry, the activation of cyclopropanes by transition metals is a research theme with implications for organic synthesis and homogeneous catalysis. Being highly strained, cyclopropanes are prone to oxidative addition to transition metal complexes. The resulting metallacycles are susceptible to a variety of reactions. These reactions are rare examples of C-C bond activation. The rarity of C-C activation processes has been attributed to Steric effects that protect C-C bonds. Furthermore, the directionality of C-C bonds as compared to C-H bonds makes orbital interaction with transition metals less favorable. Thermodynamically, C-C bond activation is more favored than C-H bond activation as the strength of a typical C-C bond is around 90 kcal per mole while the strength of a typical unactivated C-H bond is around 104 kcal per mole.
Heterobimetallic catalysis is an approach to catalysis that employs two different metals to promote a chemical reaction. Included in this definition are cases where: 1) each metal activates a different substrate, 2) both metals interact with the same substrate, and 3) only one metal directly interacts with the substrate(s), while the second metal interacts with the first.

The Mizoroki−Heck coupling of aryl halides and alkenes to form C(sp2)–C(sp2) bonds has become a staple transformation in organic synthesis, owing to its broad functional group compatibility and varied scope. In stark contrast, the palladium-catalyzed reductive Heck reaction has received considerably less attention, despite the fact that early reports of this reaction date back almost half a century. From the perspective of retrosynthetic logic, this transformation is highly enabling because it can forge alkyl–aryl linkages from widely available alkenes, rather than from the less accessible and/or more expensive alkyl halide or organometallic C(sp3) synthons that are needed in a classical aryl/alkyl cross-coupling.
In organic chemistry, the Conia-ene reaction is an intramolecular cyclization reaction between an enolizable carbonyl such as an ester or ketone and an alkyne or alkene, giving a cyclic product with a new carbon-carbon bond. As initially reported by J. M. Conia and P. Le Perchec, the Conia-ene reaction is a heteroatom analog of the ene reaction that uses an enol as the ene component. Like other pericyclic reactions, the original Conia-ene reaction required high temperatures to proceed, limiting its wider application. However, subsequent improvements, particularly in metal catalysis, have led to significant expansion of reaction scope. Consequently, various forms of the Conia-ene reaction have been employed in the synthesis of complex molecules and natural products.



















